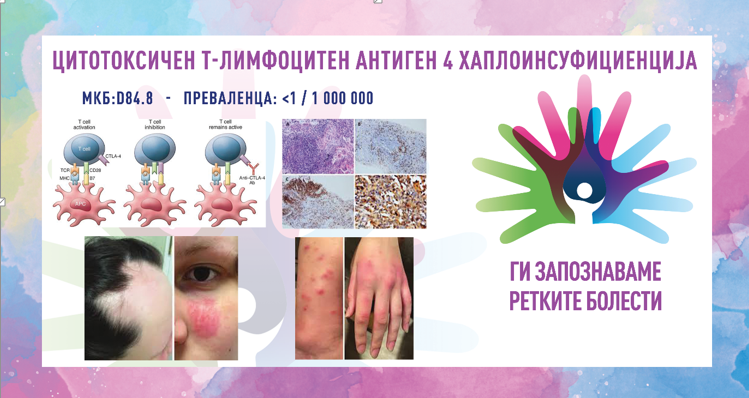
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
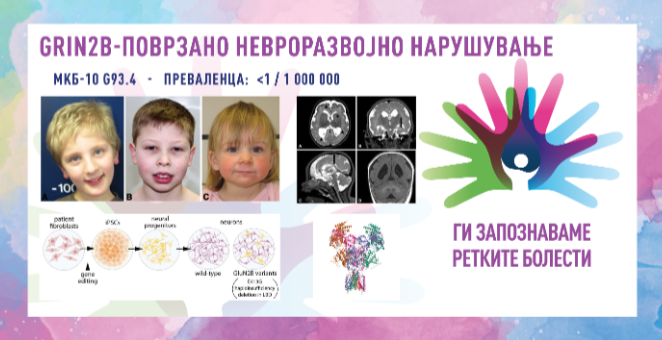
GRIN2B-ПОВРЗАНО НЕВРОРАЗВОЈНО НАРУШУВАЊЕ/GRIN2B-RELATED NEURODEVELOPMENTAL DISORDER
MKБ-10 G93.4
ПРЕВАЛЕНЦА – помалку од 1/100.000 население.
ВОВЕД
GRIN2B-поврзаното невроразвојно нарушување е исклучително ретка генетска болест со благо до изразено заостанување во развојот и интелектуално засегање кај сите пациенти. Кај одреден број пациенти присутни се и епилепсија, аутизам, нарушување на однесувањето, микроцефалија (мала глава), засегање на мускулите, меѓу кои и хипотонија и засегање на центарот за вид во мозокот. Со помош на магнетна резонанца на мозок можат да се откријат недоволно развиените делови на мозокот. Податоците евалуирани до 2022 година посочуваат дека во светот се регистрирани околу 100 пациенти со GRIN2B-поврзано невроразвојно нарушување, но се смета дека бројот е поголем бидејќи не се пријавени сите случаи.
ЕТИОЛОГИЈА
Мутациите на 12-тиот хромозом на генот GRIN2B предизвикува GRIN2B-поврзано невроразвојно нарушување. Улогата на овој ген е создавање на протеин GluN2B во невроните на мозокот. Неговата улога е во склоп на една специјализирана структура на површината на невроните која се нарекува NMDA-рецептор. Овој тип рецептори има улога во развојот на мозокот, неговата пластичност, при учење и при помнење. Некои генски мутации резултираат со создавање на нефункционален GluN2B-протеин или пак протеинот воопшто не се произведува. Кога количеството на овој протеин е намалено или воопшто го нема, тогаш се намалува и бројот на функционални NMDA-рецептори. Заболувањето се наследува автосомно-доминантно што значи дека болеста се манифестира со наследување на само една мутирана копија на овој ген. Во семејството со заболено дете вообичаено не се регистрира претходно присуство на друг заболен член или мутација кај родителите, што значи дека мутацијата настанала во процесот на репродукција.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Како што е напоменато, овие пациенти имаат лесно до тешко интелектуално засегање и забавен развој на говорот и моториката (на пр.: седење и одење). Некои индивидуи никогаш не прозборуваат, ниту пак имаат можност да проодат. Повеќето пациенти имаат хипотонија (мускулна слабост) којашто го усложнува развојот на фината и груба моторика, а го отежнува и голтањето. Може да се јави и спастицитет (згрченост) на мускулите. Повторувачките напади (епилепсија) се присутни кај околу 50 % од заболените, а кај околу 25% од заболените, се јавува аутизам, кој се карактеризира со нарушена комуникација со околината и социјална интеракција. Пациентите можат да бидат хиперактивни, импулсивни, лесно да им се одвлекува вниманието. Поретко се сретнуваат структурни абнормалности на мозокот. Доколку има таква абнормалност, малата глава (микроцефалија), нарушувањето во видот и неволевите движења на мускулите би биле знак за нешто такво. Други невролошки симптоми и знаци се церебрална парализа, хипертонија на мускулите, голема глава (макроцефалија). Во однос на дијагностицирањето не постојат типични знаци кои можат да го потврдат нарушувањето само врз основа на клиничките карактеристики. Потребно е фенетско тестирање за да се дијагностицира нарушувањето поврзано со GRIN2B, a oд голема помош можат да бидат и некои дополнителни тестови, и тоа: електроенцефалограм – се прави со цел да се пронајдат докази за абнормална активност на мозокот и нападите и МРИ – се детектираат промени во структурата на мозокот и приближно 15% од поединците кои го имаат нарушувањето, имаат екстензивна полимикрогирија.
ПОСЛЕДИЦИ
Хипотонијата влијае на способноста на детето да се исхранува, па имаат потешкотии при цицање, џвакање и голтање, а тоа пак резултира со неухранетост. Во одредени случаи може да се постави и назогастрична сонда за поуспешно хранење (сондата се поставува преку носот, грлото и хранопроводот во желудникот). Некои деца имаат рефлукс (враќање на содржината од желудникот во хранопроводот) поради што родителите треба да внимаваат во која позиција се поставени децата при хранење и спиење. Претходно наведените проблеми можат да перзистираат, но можат и да се повлечат до одреден степен. Во однос на одењето и стоењето, некои деца имаат тенденција да проодат и да научат да седат, но за оние кај кои тоа не е случај, се користат помагала за одење и инвалидски колички. Многу мал дел од пациентите покажуваат знаци на развојна регресија, при која привремено ја губат можноста за седење, лазење и одење. Постојат и пациенти кај кои има неволеви, неочекувани и повторувачки движења/контракции на дел од мускулатурата на телото.
ТРЕТМАН
Не постои лек за оваа болест, но раната дијагноза ќе значи соодветно следење и симптоматска терапија (физикална терапија, работна терапија, работа со логопед и дефектолог) во текот на животот. Децата можат да посетуваат логопед и други терапевти за надминување на проблемите со хранењето. Се препорачува генетско советување во зависност од резултатите од генетските тестирања на семејството. Согласно претходно наведеното, пациентите се следат од страна на мултидисциплинарен тим на лекари и соработници – педијатар, офталмолог, невролог, медицински генетичар, физијатар, логопед, физиотерапевт.
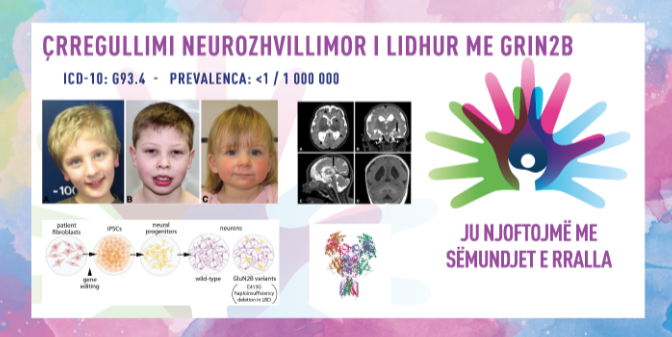
ÇRREGULLIMI NEUROZHVILLIMOR I LIDHUR ME GRIN2B/GRIN2B-RELATED NEURODEVELOPMENTAL DISORDER
ICD-10 G93.4
PREVALENCA – Prevalenca është më pak se 1 në 100,000 individë.
HYRJE
Çrregullimi neurozhvillimor i lidhur me GRIN2B është sëmundje gjenetike jashtëzakonisht e rrallë, me vonesë të lehtë deri në të rëndë në zhvillim dhe pengesa intelektuale te të gjithë pacientët. Te numër i caktuar i pacientëve mund të jenë të pranishme edhe epilepsia, autizmi, çrregullime të sjelljes, mikrocefalia (kokë e vogël), dëmtime të muskujve, mes të cilave edhe hipotonia dhe probleme me qendrën vizuale në tru. Përmes rezonancës magnetike (MRI) të trurit mund të zbulohen pjesët jo mjaftueshëm të zhvilluara të trurit. Sipas të dhënave të vlerësuara deri në vitin 2022, rreth 100 pacientë me këtë çrregullim janë regjistruar në botë, megjithëse numri i vërtetë mendohet të jetë më i lartë për shkak të rasteve të paregjistruara.
ETIOLOGJIA
Mutacionet në kromozomin e 12-të në gjenin GRIN2B shkaktojnë çrregullim neurozhvillimor të lidhur me GRIN2B. Roli i këtij gjeni është prodhimi i proteinës GluN2B në neuronet e trurit. Roli i tij është pjesë e një strukture të specializuar në sipërfaqen e neuroneve të quajtur receptor NMDA. Këta receptorë luajnë rol në zhvillimin e trurit, plasticitetin e tij, mësimin dhe kujtesën. Disa mutacione gjenetike shkaktojnë prodhim të një proteine GluN2B jo funksionale ose proteina aspak nuk prodhohet. Kur sasia e kësaj proteine është e reduktuar ose plotësisht mungon, atëherë numri i receptorëve NMDA funksionalë zvogëlohet.
Sëmundja trashëgohet në mënyrë autosomale-dominante, që do të thotë se sëmundja shfaqet me trashëgimin e vetëm një kopje të mutuar të këtij gjeni. Zakonisht, në familjet me fëmijë të sëmur nuk regjistrohet prania paraprake e ndonjë anëtari tjetër të sëmurë në familje ose mutacione te prindërit, që domethënë se mutacioni ka ndodhur gjatë procesit të riprodhimit.
SIMPTOMAT DHE DIAGNOSTIKIMI
Siç u përmend, këta pacientë kanë pengesa intelektuale nga të lehta deri në të rënda dhe zhvillim të ngadalësuar të të folurit dhe motorikës (p.sh., të ulen ose të ecin). Disa individë nuk arrijnë kurrë të flasin ose të ecin. Shumica e pacientëve kanë hipotoni (dobësi muskulore) që ndikon në zhvillimin e motorikës së imët dhe të ashpër, ndërsa gjithashtu e vështirëson gëlltitjen. Mund të shfaqet edhe spasticitet (tkurrje) i muskujve. Kriza të përsëritëshme (epilepsi) janë të pranishme te rreth 50% e të prekurve, ndërsa te rreth 25% e të sëmurëve paraqitet autizëm, i cili karakterizohet nga vështirësi në komunikim me rrethinën dhe ndërveprim social. Pacientët mund të jenë hiperaktivë, impulsivë dhe u shpërqendrohet lehtësisht vëmendja. Më rrallë hasen anomali strukturore të trurit. Për sa kohë që ka abnormalitete të tilla, kokë e vogël (mikrocefalia), çrregullime të shikimit dhe lëvizje të pavullnetshme të muskujve, do ishin shenjë për diçka të tillë. Simptoma dhe shenja të tjera neurologjike përfshijnë paralizën cerebrale, hipertoninë muskulore dhe kokë e madhe (makrocefali). Sa i përket diagnozës nuk ka shenja tipike që mund të konfirmojnë çrregullimin vetëm sipas karakteristikave klinike. Është i nevojshëm testimi gjenetik për të diagnostikuar çrregullimin e lidhur me GRIN2B, ndërsa ndihmë të madhe mund të ofrojnë edhe disa teste shtesë, siç janë: elektroencefalogrami – bëhet me qëllim të zbulohen dëshmi të aktivitetit abnormal të trurit dhe krizave; MRI e trurit – detektohen ndryshime në strukturën e trurit dhe përafërsisht 15% e individëve me këtë çrregullim kanë polimikrogiri të zgjeruar.
PASOJAT
Hipotonia ndikon në aftësinë e fëmijës për t'u ushqyer, prandaj kanë vështirësi në thithje, përtypje dhe gëlltitje, ndërsa kjo rezulton në kequshqyerje. Në disa raste, mund të vendoset një sondë nazogastrike për ushqyerje më efektive (sonda kalon përmes hundës, fytit dhe ezofagut në stomak). Disa fëmijë përjetojnë refluks (rikthim të përmbajtjes së stomakut në ezofag), prandaj prindërit duhet të kenë kujdes në çfarë pozicioni janë fëmijët gjatë ushqyerjes dhe gjumit. Problemet e lartpërmendura mund të perzistojnë ose të përmirësohen pjesërisht. Sa i përket ecjes dhe qëndrimin, disa fëmijë kanë tendencë të ecin dhe të mësojnë të ulen, por për ata që nuk e arrijnë këtë, përdoren pajisje mbështetëse për ecje dhe karroca me rrota. Një pjesë shumë e vogël e pacientëve paraqesin shenja të regresionit zhvillimor, gjatë cilit përkohësisht e humbin aftësinë e uljes, zvarritjes dhe ecjes. Ka pacientë të cilët kanë lëvizje të pavullnetshme, të papritura dhe të përsëritura të muskujve të trupit.
TRAJTIMI
Nuk ka kurë për këtë sëmundje, por diagnoza e hershme ndihmon në ndjekjen e duhur dhe terapi simptomatike gjatë jetës (terapi fizike, terapi profesionale, punë me logoped dhe defektolog). Fëmijët mund të vizitojnë defektologë dhe terapistë të tjerë për tejkalimin e problemeve të ushqyerjes. Konsultimi gjenetik rekomandohet në varësi të rezultateve të testeve gjenetike të familjarëve. Si përfundim, pacientët ndiqen nga një ekip multidisiplinor mjekësh dhe punonjësish mjekësorë – pediatër, oftalmolog, neurolog, gjenetist mjekësor, fiziatër, logoped dhe fizioterapist.