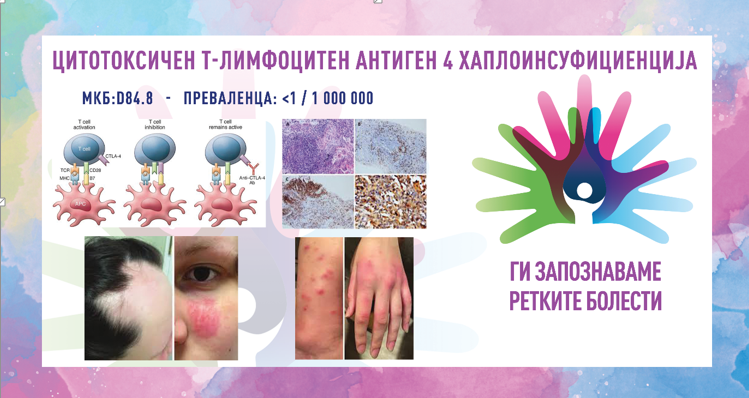
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
НИЈМЕГЕНОВ СИНДРОМ НА РАСКИНУВАЊЕ/ НИЈМЕГЕНОВ СИНДРОМ НА КРШЕЊЕ/ NIJMEGEN BREAKAGE SYNDROME
MKБ-10 Q87.8 (Q90.9, D81.9)
ПРЕВАЛЕНЦА – непозната, иако постојат податоци дека преваленцата кај новороденчињата со источнословенско потекло (Белорусија, Украина, Русија и Латвија) била проценета на 1/1.000.000 живородени, додека преваленцата во западнословенскиот регион (Полска, Словачка и Чешка) се проценува на 1/330.000.
ВОВЕД
Нијмегенов синдром на раскинување (НСР) е редок синдром на генетска, хромозомска нестабилност која најверојатно настанува како резултат на дефект во механизмот за поправка на ДНК со двојна врска и/или механизмот за анелирање за поправка на прекинот на двојната врска во ДНК. Ова автосомно рецесивно нарушување за првпат било опишано кај холандски пациенти кои најверојатно биле потомци на полски или чешки граѓани кои емигрирале во Холандија за време на 1600-тите по 30-годишната војна бидејќи поголемиот дел од пациентите со НСР всушност живеат во Полска и Чешка. При раѓањето, новороденчињата се манифестираат со микроцефалија, дисморфни црти на лицето кои стануваат позабележителни со возраста, одложување на растот, повторливи синопулмонални инфекции и екстремно висока фреквенција на малигни тумори во раната возраст. Моделот на наследување е автосомно рецесивен.
ЕТИОЛОГИЈА
Нијмегеновиот синдром на кршење е предизвикан од мутации во генот NBN (8q21-q24), конкретно во рамките на егзоните 6-10, кои доведуваат до делумно функционални скратени фрагменти на нибрин, генски производ вклучен во поправка на прекините на ДНК со двојна врска. Над 90 % од пациентите од словенско потекло се хомозиготни.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
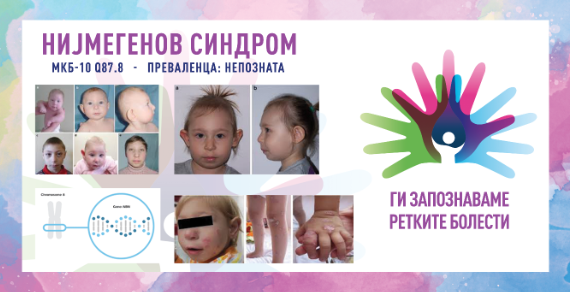
Клиничките манифестации не се типични и можат да се разликуваат според степенот на сериозност. Главните знаци се микроцефалија, присутна при раѓање која напредува со возраста, дисморфни црти на лицето (истакната средина нагласена со наведнато чело и повлекување на мандибулата), блага ретардација на растот и, кај женските пациенти, се јавува честа предвремена оваријална инсуфициенција. Пријавени се различни вродени аномалии, вклучително и во централен нервен систем (хидроцефалија, шизенцефалија, арахноидни цисти), респираторен тракт (расцеп на усна/непце, хоанална атрезија), урогенитален систем (потковица бубрег, ектопични/дистопични бубрези, хипоспадија, хипоспадија, крипторија), благи скелетни аномалии (пред- и постаксијална полидактилија, хипопластичен или двоен палец, клинодактилија на петтиот прст). Често се забележуваат дамки „café au lait“ и/или витилиго, а кај некои пациенти се јавуваат повеќе пигментирани нерви. Другите главни манифестации вклучуваат имунолошки дефицит со рекурентни инфекции на респираторниот тракт, силна предиспозиција за малигни тумори (претежно лимфоидни, но се појавуваат и цврсти тумори) и радиочувствителност. До 20-годишна возраст, над 40 % од пациентите развиваат малигно заболување и се склони кон развој на секундарни малигни тумори. Когнитивниот развој е блиску до просекот (нормален/граничен) во детството и предучилишната возраст, но интелектуалните вештини постепено опаѓаат со возраста (од благ до умерен).
Дијагнозата се заснова на клиничките манифестации и се потврдува или со секвенционирање на еден ген (обично за словенските популации) или со мултигенски панели за секвенционирање на следната генерација. Онаму каде што генетското тестирање е недостапно, дијагнозата може да биде поддржана со докази за хромозомска нестабилност (спонтана и индуцирана), зголемена клеточна чувствителност на јонизирачко зрачење, ин витро, комбинирана имунодефициенција и целосно отсуство на нибрин со целосна должина. Семејната историја (малигни заболувања, микроцефалија или хидроцефалија, рана смрт на брат или сестра) исто така може да ја дефинира дијагнозата. Особено важно е да се исклучат диференцијалните дијагнози, како што се Fanconi-анемија, LIG4-синдром, Cernunnos-XLF-дефицит, нарушување слично на NBS, нарушување слично на атаксија-телеангиектазија и Блумов синдром
ПОСЛЕДИЦИ
Прогнозата е лоша, со малигнитетот како главна причина за смрт. Пациентите кои развиле лимфом или леукемија починале од прогресија на болеста, релапси и секундарни малигни тумори. Корисниот ефект на HSCT врз долгорочното преживување е потврден во неодамнешните студии.
ТРЕТМАН
За жал, и за овој синдром, како и повеќето, не постои специфична терапија. Од големо значење е раната дијагноза со цел да се избегнат тешки рекурентни инфекции, непотребна изложеност на зрачење за дијагностички цели и негативни ефекти од радиотерапијата на туморите. Родителите на засегнатото дете се задолжителни носители на мутациите на НСК и на тој начин, за секоја бременост, постои 25 % ризик за потомството да ја наследи болеста. На носителите на словенска основачка мутација треба да им се понуди следење на ракот, особено на дојката кај жените и на простатата кај мажите. На пациентите им е потребен мултидисциплинарен третман и долгорочно следење (малигнитет, имунодефициенција, раст, хипергонадотропен хипогонадизам кај жени). Следењето на имунолошкиот систем е исклучително важно во текот на животот; неопходни се специфични начини и типови на имунизација (се препорачуваат ацелуларни вакцини). Во случај на лимфоидна малигност, се препорачува трансплантација на хематопоетски матични клетки (HSCT) по првата целосна ремисија. Во поглед на припадничките на женскиот пол, се нуди следење на напредокот од 12-годишна возраст (ендокринолог/гинеколог) и хормонска терапија за соодветната возраст.

SINDROMA E THYERJES NIJMEGEN/ NIJMEGEN BREAKAGE SYNDROME
ICD-10 Q87.8 (Q90.9, D81.9)
PREVALENCA – e panjohur, megjithëse ka të dhëna që prevalenca te të porsalindurit me origjinë sllave lindore (Bjellorusia, Ukraina, Rusia dhe Letonia) është vlerësuar në 1/1.000.000 lindje të gjalla, ndërsa prevalenca në rajonin sllav perëndimor (Poloni, Sllovaki dhe Republika Çeke) u vlerësua në 1/330.000.
HYRJE
Sindroma e thyerjes Nijmegen (NSR) është sindromë e rrallë e paqëndrueshmërisë gjenetike, kromozomale që ka shumë të ngjarë të jetë rezultat i një defekti në mekanizmin e riparimit të thyerjes së dyfishtë të ADN-së ose mekanizmit të pjekjes për riparimin e thyerjes së dyfishtë të ADN-së. Ky çrregullim autosomal recesiv u përshkrua për herë të parë në pacientët holandezë, të cilët me gjasë ishin pasardhës të qytetarëve polakë ose çekë që emigruan në Holandë gjatë viteve 1600 pas Luftës 30-vjeçare, sepse shumica e pacientëve me NSR jetojnë në Poloni dhe Republikën Çeke. Në lindje, foshnjat prezantohen me mikrocefali, tipare dismorfike të fytyrës që bëhen më të dukshme me moshën, ngadalësim të rritjes, infeksione të përsëritura sinopulmonare dhe një frekuencë jashtëzakonisht të lartë të sëmundjeve malinje në moshë të hershme. Modeli i trashëgimisë është autosomik recesiv.
ETIOLOGJIA
Sindroma e thyerjes Nijmegen shkaktohet nga mutacionet në gjenin NBN (8q21-q24), veçanërisht brenda ekzoneve 6-10, të cilat çojnë në fragmente të cunguara pjesërisht funksionale të nibrinës, një produkt gjeni i përfshirë në riparimin e thyerjeve të dyfishtë të ADN-së. Mbi 90% e pacientëve me origjinë sllave janë homozigotë.
SIMPTOMAT DHE DIAGNOSTIKIMI
Manifestimet klinike nuk janë tipike dhe mund të ndryshojnë në ashpërsi. Shenjat kryesore janë mikrocefalia e pranishme në lindje e cila përparon me kalimin e moshës, tiparet dismorfike të fytyrës (vija e mesme e theksuar e theksuar nga një ballë e përkulur dhe tërheqja e mandibulës), vonesa e lehtë e rritjes dhe në pacientët femra ndodh shpesh dështimi i parakohshëm i vezoreve. Janë raportuar një sërë anomalish kongjenitale, duke përfshirë në sistemin nervor qendror (hidrocefalus, skizencefali, kista arachnoid), traktin respirator (kuzë e çarë/qiellzë, atrezi choanale), sistemin urogjenital (veshka patkoi, veshka ektopike/distopike, hipospadia, kriptoria), anomalitë e lehta skeletore (polidaktilia para dhe postaksiale, hipoplastike ose gishti i dyfishtë, klinodaktilia e gishtit të pestë). Shpesh shihen njolla kafeje ose vitiligo dhe disa pacientë zhvillojnë nerva të shumëfishtë të pigmentuar. Manifestime të tjera kryesore përfshijnë mungesën e imunitetit me infeksione të përsëritura të traktit respirator, një predispozitë të fortë ndaj tumoreve malinje (kryesisht limfoide, por shfaqen edhe tumore të ngurta) dhe ndjeshmëri ndaj radios. Deri në moshën 20 vjeçe, mbi 40% e pacientëve zhvillojnë një sëmundje malinje dhe janë të prirur për zhvillimin e tumoreve malinje dytësore. Zhvillimi kognitiv është afër mesatares (normale/kufitare) në fëmijëri dhe parashkollor, por aftësitë intelektuale bien gradualisht me kalimin e moshës (i butë deri në mesatar).
Diagnoza bazohet në manifestimet klinike dhe konfirmohet ose nga sekuenca me një gjen (zakonisht për popullatat sllave) ose nga panelet e sekuencës së gjeneratës së ardhshme multigjenike. Aty ku testimi gjenetik nuk është i disponueshëm, diagnoza mund të mbështetet nga dëshmitë e paqëndrueshmërisë kromozomale (spontane dhe të induktuar), ndjeshmërisë së rritur qelizore ndaj rrezatimit jonizues, in vitro, mungesës së imunitetit të kombinuar dhe mungesës së plotë të nibrinës me gjatësi të plotë. Historia familjare (malinje, mikrocefali ose hidrocefalus, vdekja e hershme e një vëllai ose motre) gjithashtu mund të përcaktojë diagnozën. Është veçanërisht e rëndësishme të përjashtohen diagnozat diferenciale si anemia Fanconi, sindroma LIG4, mungesa e Cernunnos-XLF, çrregullimi i ngjashëm me NBS, çrregullimi i ngjashëm me ataksi-teleangiektaze dhe sindroma e Bloom-it.
PASOJAT
Prognoza është e keqe, ku shkaku kryesor i vdekjes është malinjiteti. Pacientët që zhvilluan limfomën ose leukeminë vdiqën nga progresi i sëmundjes, relapsat dhe sëmundjet malinje sekondare. Efekti i dobishëm i HSCT në mbijetesën afatgjatë është konfirmuar në studimet e fundit.
TRAJTIMI
Fatkeqësisht për këtë syndrome si shumica e tyre nuk ka terapi specifike. Diagnoza e hershme ka një rëndësi të madhe për të shmangur infeksionet e rënda të përsëritura, ekspozimin e panevojshëm ndaj rrezatimit për qëllime diagnostikuese dhe efektet negative të radioterapisë së tumorit. Prindërit e fëmijës së prekur janë bartës të detyrueshëm të mutacioneve të NSC dhe kështu, për çdo shtatzëni, ekziston një rrezik 25% që pasardhësit të trashëgojnë sëmundjen. Bartësve të mutacionit themelues sllav duhet t'u ofrohet ndjekja e kancerit, veçanërisht e gjirit te femrat dhe e prostatës te meshkujt. Pacientët kërkojnë trajtim multidisiplinar dhe ndjekje afatgjatë (malinjiteti, imunodefiçenca, rritja, hipogonadizmi hipergonadotropik te gratë). Monitorimi i sistemit imunitar është jashtëzakonisht i rëndësishëm gjatë gjithë jetës; metodat dhe llojet specifike të imunizimit janë të nevojshme (rekomandohen vaksinat acelulare). Në rast malinje limfoide, transplantimi i qelizave staminale hematopoietike (HSCT) rekomandohet pas faljes së parë të plotë. Për femrat ofrohet monitorim nga mosha 12 vjeçe (endokrinolog/gjinekolog) dhe terapi hormonale e përshtatshme për moshën.