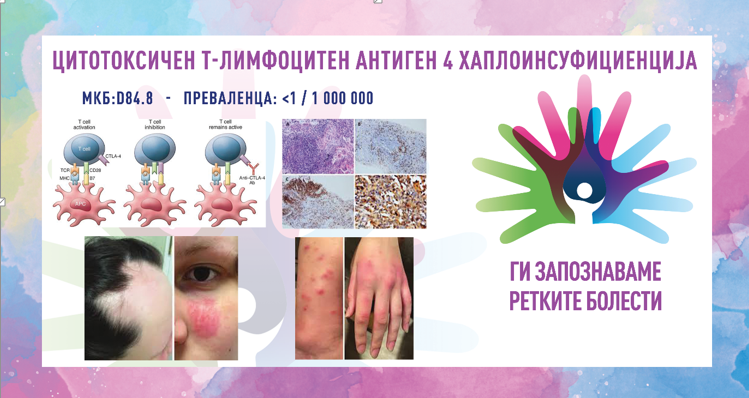
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
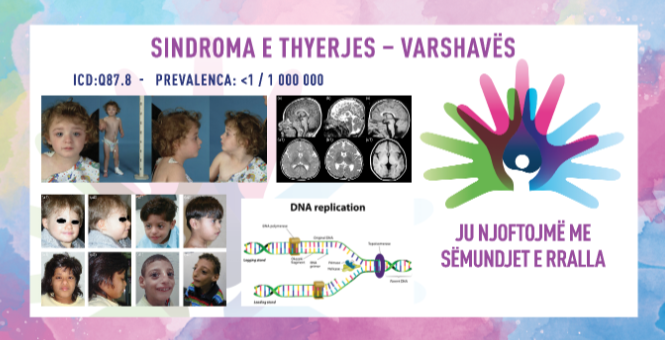
СИНДРОМ НА КРШЕЊЕ – ВАРШАВА / WARSAW BREAKAGE SYNDROME
MKБ-10 Q87.8
ПРЕВАЛЕНЦА – забележана кај помалку од 100 живородени деца, особено кај Ашкеназите.
ВОВЕД
Синдромот на кршење – Варшава е генетски, мултисистемски, дисморфен синдром кој се карактеризира со пред- и постнатално ограничување на растот, микроцефалија, блага до тешка интелектуална попреченост, сензоневрален губиток на слухот со кохлеарни абнормалности и фацијален дисморфизам (со мало и издолжено лице, бифронтално стеснување, кратки нопични, мали нари, диспластични уши и краток врат). Дополнително, пациентите родени со оваа состојба доживуваат оштетувања на слухот, умерени до значителни доцнења во развојот, скелетни нарушувања и нарушувања на срцето (40%), како и нарушувања на различни телесни системи, како што се бубрежните и урогениталните системи, а кај некои пациенти, тестовите на крвта укажуваат на зголемена кршливост на хромозомите (некои фетуси умираат во матката). Синдромот за првпат бил опишан во 2010 година, а до 2018 година биле пријавени помалку од десет случаи.
ЕТИОЛОГИЈА
Синдромот е предизвикан од мутации во генот DDX11, кој дава инструкции за создавање ензим наречен ChlR1. Овој ензим функционира како хеликаза. Хеликазите се ензими кои се прикачуваат (се врзуваат) за ДНК и привремено ги одмотуваат двете спирални нишки (двојна спирала) на молекулата на ДНК. Ова одмотување е неопходно за реплицирање на ДНК во подготовка за клеточна делба и за поправка на оштетената ДНК и на сите грешки што се прават при процесот на реплицирање. Дополнително, откако ќе се реплицира ДНК, ChlR1 игра улога во обезбедувањето на правилно одвојување на секој хромозом за време на клеточната делба. Со поправка на грешките во ДНК и обезбедување на правилна репликација на ДНК, ензимот ChlR1 е вклучен во одржувањето на стабилноста на генетските информации на клетката. Мутациите на генот DDX11 сериозно ја намалуваат или целосно ја елиминираат активноста на ензимот ChlR1. Како резултат на тоа, ензимот не може да се поврзе со ДНК и не може да ги отвори нишките на ДНК за да помогне во репликацијата и поправката на ДНК. Недостатокот на функционален ChlR1 ја нарушува клеточната делба и доведува до акумулација, односно оштетување. Ова оштетување на ДНК може да се појави како прекини во ДНК, давајќи го името на состојбата. Не е јасно како овие проблеми во одржувањето на ДНК доведуваат до специфични абнормалности карактеристични за овој синдром на кршење. Нарушувањето следи автосомно рецесивен модел на наследување, што значи дека и двете копии на генот мораат да бидат мутирани за да се манифестира синдромот.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Клиничките карактеристики кај лицата кои го имаат синдромот на кршење вклучуваат:
тешка пред- и пост натална ретардација на растот – вообичаен низок раст;
задоцнување во развојот – когнитивниот и моторниот развој можат да бидат одложени, а честа е појавата на интелектуална попреченост;
краниофацијални абнормалности – широк, рамен назален мост, краток и рамен филтрум, мало и издолжено лице, истакнати образи, релативно голема уста и големи уши;
скелетни абнормалности – хипопластични палци, сколиоза и други проблеми со скелетот, клинодактилија на петтиот прст, синдактилија на вториот и третиот прст, мали палци;
имунолошки дефицит – зголемена подложност на инфекции поради абнормалности на имунолошкиот систем;
наоди на кожата – може да се појави единечна палмарна бразда, сува кожа, осип и абнормална пигментација на кожата;
невролошки проблеми – напади, хиперактивност и други невролошки дефицити.
Дијагностицирањето започнува со комбинација на клинички карактеристики, семејна историја и резултати од генетското тестирање кое може да помогне во идентификувањето на мутации во генот BML. Друг главен тест е анализата на хромозомското кршење, преку кој се открива зголемено кршење на хромозомите во култивираните лимфоцити. Х-зраците, компјутеризираната томографија или МРИ можат да помогнат за проценка на скелетните абнормалности или други структурни дефекти. При дијагностицирањето треба да се внимава на диференцијалните дијагнози бидејќи неколку генетски и метаболитички нарушувања можат да посочуваат на основните карактеристики на овој синдром. Диференцијалните дијагнози вклучуваат:
Блумов синдром – слична хромозомска нестабилност и ретардација во растот, се разликува во имунолошкиот дефицит и специфичните црти на лицето;
Фанкониева анемија – се манифестира со хромозомска нестабилност, но вклучува различни генетски мутации;
Синдром на Швахман–Дијамант – се карактеризира со дисфункција на коскената срцевина, инсуфициенција на панкреасот и ретардација на растот, што може да се преклопува со некои карактеристики на синдромот на кршење;
цистична фиброза – иако првенствено станува збор за респираторно нарушување, исто така може да доведе до ретардација на растот и имунолошки проблеми.
ПОСЛЕДИЦИ
Прогнозата за лицата засегнати со овој синдром варира врз основа на сериозноста на клиничките манифестации. Очекуваното траење на животот на лицата со овој синдром на кршење генерално е намалено поради зголемениот ризик од инфекции, рак и други компликации. Сепак, со соодветна помош и грижа, многу поединци можат да живеат до доба на зрелоста, иако можат да доживеат и старост како кај останатите поединци.
ТРЕТМАН
Генерално, не постои лек за синдромот на кршење, но поддршката и некои одредени третмани можат да бидат од помош. Специфичните третмани вклучуваат:
поддршка за раст и развој – програми за рана интервенција за доцнење во развојот, говорна терапија и физикална терапија;
поддршка на имунолошкиот систем – профилактички антибиотици и други третмани за управување со инфекции поради имунолошки дефицит;
нега на скелетот – може да вклучи ортопедска нега за сколиоза или малформации на екстремитетите;
управување со напади – антиконвулзивни лекови за контрола на нападите, доколку е потребно;
редовно следење – поради ризикот од малигни тумори, лицата со синдромот на кршење треба да подлежат на редовен мониторинг за рак;
скрининг на носител – семејствата можат да имаат корист од генетско советување
и скрининг за да утврдат дали носат мутација во генот BLM;
проценка на ризик – ако и двајцата родители се носители на BLM-мутацијата, постои 25% ризик со секоја бременост детето да биде засегнато од нарушувањето;
репродуктивни опции – семејствата можат да разгледуваат и репродуктивни опции, како што е ин витро оплодувањето (ИВФ) со предимплантациско генетско тестирање (PGT) за да се намали ризикот од заболено дете.
SINDROMA E THYERJES – VARSHAVËS / WARSAW BREAKAGE SYNDROME
ICD-10 Q87.8
PREVALENCA – vërehet në më pak se 100 lindje të gjalla, veçanërisht në mesin e Ashkenazimëve.
HYRJE
Sindroma Varshavë është sindromë gjenetike, shumësistemike, dismorfike e karakterizuar nga kufizimi i rritjes para dhe pas lindjes, mikrocefali, paaftësi intelektuale e lehtë deri në të rëndë, humbje dëgjimi sensorineural me anomali kokleare dhe dismorfizëm të fytyrës (me fytyrë të vogël dhe të zgjatur, ngushtim bifrontal, hundë të shkurtra, vrima të vogla të hundës, veshë displastikë dhe qafë të shkurtër). Përveç kësaj, pacientët e lindur me këtë gjendje përjetojnë dëmtim të dëgjimit, vonesa të moderuara deri në të konsiderueshme të zhvillimit, anomali skeletore dhe kardiake (40%), si dhe çrregullime të sistemeve të ndryshme të trupit, si sistemet renale dhe urogjenitale, dhe në disa pacientë, analizat e gjakut. tregojnë rritjen e brishtësisë së kromozomeve (disa fetuse vdesin në mitër). Sindroma u përshkrua për herë të parë në vitin 2010 dhe deri në vitin 2018, ishin raportuar më pak se dhjetë raste.
ETIOLOGJIA
Sindroma shkaktohet nga mutacionet në gjenin DDX11, i cili jep udhëzime për krijimin e një enzime të quajtur ChlR1. Kjo enzimë funksionon si një helikazë. Helikazat janë enzima që lidhen (lidhen) me ADN-në dhe zbërthejnë përkohësisht dy vargjet spirale (spiralja e dyfishtë) të molekulës së ADN-së. Ky zbërthim është i nevojshëm për replikimin e ADN-së në përgatitje për ndarjen e qelizave dhe për riparimin e ADN-së së dëmtuar dhe çdo gabimi të bërë gjatë procesit të replikimit. Për më tepër, pasi ADN-ja të riprodhohet, ChlR1 luan një rol në sigurimin e ndarjes së duhur të secilit kromozom gjatë ndarjes së qelizave. Duke riparuar gabimet e ADN-së dhe duke siguruar replikimin e duhur të ADN-së, enzima ChlR1 përfshihet në ruajtjen e stabilitetit të informacionit gjenetik të qelizës. Mutacionet në gjenin DDX11 reduktojnë ose eliminojnë
plotësisht aktivitetin e enzimës ChlR1. Si rezultat, enzima nuk mund të lidhet me ADN-në dhe nuk mund të hapë fijet e ADN-së për të ndihmuar në riprodhimin dhe riparimin e ADN-së. Mungesa e ChlR1 funksionale prish ndarjen qelizore dhe çon në akumulim ose dëmtim. Ky dëmtim i ADN-së mund të shfaqet si thyerje në ADN, duke i dhënë gjendjes emrin e saj. Nuk është e qartë se si këto probleme në mirëmbajtjen e ADN-së çojnë në anomalitë specifike karakteristike të kësaj sindrome të thyerjes. Çrregullimi ndjek një model autosomik recesiv të trashëgimisë, që do të thotë se të dyja kopjet e gjenit duhet të mutohen në mënyrë që sindroma të shfaqet.
SIMPTOMAT DHE DIAGNOSTIKIMI
Karakteristikat klinike te individët që kanë sindromën e shtypjes përfshijnë:
vonesë të rëndë të rritjes para dhe pas lindjes – shtat i shkurtër i zakonshëm;
vonesë në zhvillim – zhvillimi kognitiv dhe motorik mund të vonohet dhe paaftësia intelektuale është e zakonshme;
anomalitë kraniofaciale – hundë e gjerë, e sheshtë, filtrum i shkurtër dhe i sheshtë, fytyrë e vogël dhe e zgjatur, faqe të theksuara, gojë relativisht e madhe dhe veshë të mëdhenj;
anomalitë të skeletit – gishtat e mëdhenj hipoplastikë, skolioza dhe probleme të tjera skeletore, klinodaktilia e gishtit të pestë, sindaktilia e gishtit të dytë dhe të tretë, gishtat e mëdhenj të vegjël;
mungesë të imunitetit – rritja e ndjeshmërisë ndaj infeksioneve për shkak të anomalive të sistemit imunitar;
gjetje të lëkurës – mund të ndodhë një rrudhë e vetme palmare, lëkurë e thatë, skuqje dhe pigmentim jonormal i lëkurës;
probleme neurologjike – konvulsione, hiperaktivitet dhe deficite të tjera neurologjike.
Diagnoza fillon me një kombinim të veçorive klinike, historisë familjare dhe rezultateve të testimit gjenetik që mund të ndihmojnë në identifikimin e mutacioneve në gjenin BML. Një tjetër test i madh është analiza e thyerjes së kromozomeve, e cila zbulon thyerje të shtuar të kromozomeve në limfocitet e kultivuara. Rrezet X, tomografia e kompjuterizuar ose MRI mund
të ndihmojnë në vlerësimin e anomalive skeletore ose defekteve të tjera strukturore. Gjatë diagnostikimit duhet t'i kushtohet vëmendje diagnozave diferenciale, sepse disa çrregullime gjenetike dhe metabolike mund të tregojnë tiparet themelore të kësaj sindrome.
Diagnozat diferenciale përfshijnë:
sindromë të Bloom – paqëndrueshmëria e ngjashme kromozomale dhe vonesa e rritjes, ndryshon në mungesë imunitare dhe tipare specifike të fytyrës;
anemi Fanconi – manifestohet me paqëndrueshmëri kromozomale, por përfshin mutacione të ndryshme gjenetike;
sindromë Shwachman–Diamond – karakterizohet nga mosfunksionimi i palcës së eshtrave, pamjaftueshmëria e pankreasit dhe ngadalësimi i rritjes, të cilat mund të mbivendosen me disa tipare të sindromës së këputjes;
fibroza cistike – megjithëse është kryesisht një çrregullim i frymëmarrjes, ajo gjithashtu mund të çojë në vonesë të rritjes dhe probleme imune.
PASOJAT
Prognoza për individët e prekur me këtë sindromë ndryshon në bazë të ashpërsisë së manifestimeve klinike. Jetëgjatësia e njerëzve me këtë sindromë të shtypjes në përgjithësi zvogëlohet për shkak të rritjes së rrezikut të infeksioneve, kancerit dhe komplikimeve të tjera. Megjithatë, me ndihmën dhe kujdesin e duhur, shumë individë mund të jetojnë deri në moshën madhore, megjithëse ata gjithashtu mund të përjetojnë pleqërinë si individë të tjerë.
TRAJTIMI
Në përgjithësi, nuk ka kurë për sindromën e Varshavës, por mbështetja dhe disa trajtime mund të jenë të dobishme. Trajtimet specifike përfshijnë:
mbështetje të rritjes dhe zhvillimit – programet e ndërhyrjes së hershme për vonesat në zhvillim, terapinë e të folurit dhe terapinë fizike;
mbështetje për sistemin imunitar – antibiotikë profilaktikë dhe trajtime të tjera për të menaxhuar infeksionet për shkak të mungesës së imunitetit;
kujdes për skeletin – mund të përfshijë kujdesin ortopedik për skoliozën ose keqformimet e gjymtyrëve;
menaxhim të konvulsioneve – medikamente antikonvulsante për të kontrolluar krizat, nëse është e nevojshme;
monitorim të rregullt – për shkak të rrezikut të tumoreve malinje, njerëzit me sindromën e Varshavës duhet t'i nënshtrohen monitorimit të rregullt për kancer;
ekzaminim të bartësit – familjet mund të përfitojnë nga këshillimi dhe kontrolli gjenetik për të përcaktuar nëse ato mbartin një mutacion në gjenin BLM;
vlerësim të rrezikut – nëse të dy prindërit janë bartës të mutacionit BLM, ekziston një rrezik 25% me çdo shtatzëni që fëmija të preket nga çrregullimi;
opsione riprodhuese – familjet gjithashtu mund të marrin në konsideratë opsionet riprodhuese, të tilla si fekondimi in vitro (IVF) me testimin gjenetik të paraimplantit (PGT) për të reduktuar rrezikun për të pasur një fëmijë të prekur.