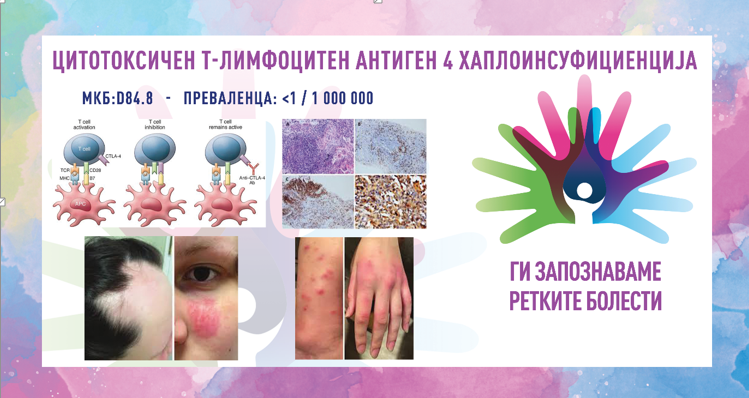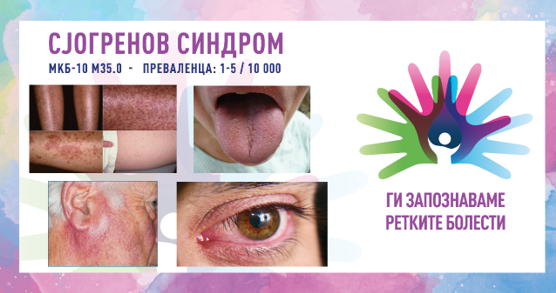
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
АЛКАПТОНУРИЈА/ БОЛЕСТ НА ЦРНА УРИНА/
ALKAPTONURIA/ BLACK URINE DISEASE
MKБ-10 Е70.2
ПРЕВАЛЕНЦА – забележана кај 1/250 000 живородени деца до 1/1 000 000 кај одредени етнички групи.
ВОВЕД
Алкаптонурија е многу ретко генетско заболување кое се наследува автосомно-рецесивно, па затоа кај многу индивидуи се јавува асимптоматски со тоа што и самите не се свесни за својата состојба сè до периодот на зрелост. Пациентите немаат доволно функционални нивоа на ензим потребен за разградување на хомогентизичната киселина, со што доаѓа до појава на темна (црна) урина. Алкаптонуријата е меѓу првите генетски нарушувања кои покажаа дека ги следат принципите на Менделевото рецесивно наследување. Историски гледано, се верува дека египетската мумија Харва е првиот клинички случај на алкаптонурија кој датира уште пред 1 500 години пр.н.е. Болеста подеднакво ги погодува и мажите и жените, иако симптомите имаат тенденција на ран развој кај машките живородени деца, а како области со зголемена фреквенција се идентификувани областите во Словачка, Германија и Доминиканска Република.
ЕТИОЛОГИЈА
Болеста се развива како последица на недостаток на генот HGD, односно недостаток на ензимот хомогентизат оксидаза, потребен за разградување на аминокиселините фенилаланин и тирозин, со што пациентите, кај кои е откриена болеста, се хомозиготни или сложено хетерозиготни за мутации кои ја губат функцијата. За едно новороденче да развие алкаптонурија, потребно е да наследи две копии од неисправниот ген HGD, притоа шансите се премногу мали, со што оваа состојба е навистина ретка. Кога има недостаток, хомогентизичната киселина се акумулира во телото и на крајот се излачува во урината, давајќи ѝ темна боја при изложување на воздух. Оштетувањето на ткивото е резултат на таложење на пигмент кој е сличен на меланин (полимеризирана форма на BQA) и кој може да предизвика бројни реакции и производство на слободни радикали, со што доаѓа до дополнително оштетување на ткивото.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Еден од најраните знаци на алкаптонуријата се темните пелени кај новороденчињата, па затоа, овој знак не смее да се пропушти или занемари, сè со цел нарушувањето да не остане незабележано. Освен темната преобоеност на урината во доба на детство, заболените, главно, не развиваат други симптоми сè до повозрасната доба од животот (вообичаено околу доцните 20-ти до раните 30-ти години) и затоа долго време не се свесни за постоењето на ова заболување кај нив. Во текот на животот, хомогентизичната киселина почнува да се депонира и таложи во различни делови од организмот, давајќи карактеристични симптоми и оштетувања, и тоа:
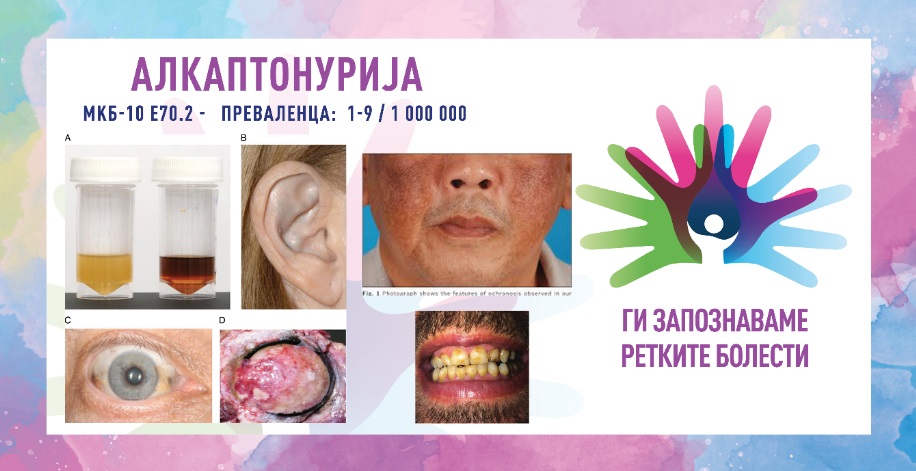
- промена на бојата на урината– најчест симптом, потемнување на урината при изложување на воздух;
- охроноза– сино-црна промена на бојата на сврзните ткива, вклучувајќи ја ᾿рскавицата и кожата; може да влијае на различни области, особено на ушите, носот склерата и ноктите;
- проблеми со зглобовите– многу пациенти доживуваат артритис, особено во поголемите зглобови, како што се рамената, колковите и колената, поради акумулацијата на пигмент во ᾿рскавицата;
- срцеви проблеми– наслагите можат да се акумулираат и во срцевите залистоци, што доведува до потенцијални кардиоваскуларни проблеми, како стенози, аритмии, срцева слабост и слично;
- камења во бубрезите– зголеменото ниво на хомогентизична киселина може да доведе до формирање на камења во бубрезите.
Бидејќи постојат повеќе диференцијални дијагнози (акутна интермитентна порфирија, ревматоиден артритис, анкилозен спондилитис, остеоартритис), потребно е земање на детална анамнеза и спроведување на неколку тестови за докажување на болеста. Клучна дијагностичка алатка е уринарниот тест, каде примерокот станува темен кога ќе се изложи на воздух. Зголемените нивоа на хомогентизична киселина можат да се потврдат преку хемиска анализа. Може да се изврши и генетско тестирање за да се идентификуваат мутациите во генот HGD, со што ќе се обезбеди дефинитивна дијагноза и потоа ќе следи генетско советување. Рендгенското снимање или МРИ можат да се користат за да се процени оштетувањето на зглобовите и да се следи прогресијата на артритисот.
ПОСЛЕДИЦИ
Очекуваниот животен век кај пациентите кои имаат алкаптонурија не е значително намален во однос на просечниот животен век, но постојаната болка може да биде повод за прогресивен функционален пад со губење на подвижноста, со што пациентите често користат помагала при движењето. Дополнително, срцевите компликации често се опасни по живот и можат да ја влошат состојбата.
ТРЕТМАН
Иако не постои лек, третманот се насочува кон ублажување на симптомите и подобрување на квалитетот на животот. Терапијата се заснова на: нестероидни антиинфламаторни лекови (НСАИЛ) кои најчесто се користат за справување со болки во зглобовите и воспаление. Измените во исхраната се корисни иако не постои специфична диета за лекување на алкаптонурија, сепак некои препораки вклучуваат намален внес на протеини, а со тоа и на тирозин и фенилаланин за да се ограничи производството на хомогентизична киселина. Физикална терапија и операција се применуваат во понапреднати фази кога имаме ограничена функција на зглобовите, кога артритисот е многу напреднат, кога имаме намалена мобилност во зглобовите или кога оштетувањето е толку големо што резултира со потреба од соодветна хируршка интервенција. Новите истражувања влеваат надеж за идни нови видови терапија и излекување на болеста. Сепак, свеста за состојбата останува од суштинско значење за рана дијагноза и ефикасна нега.
ALKAPTONURIA/
ALKAPTONURIA/ BLACK URINE DISEASE
ICD-10 Е70.2
PREVALENCA – është vërejtur te 1/250,000 fëmijë të lindur të gjallë deri në 1/1,000,000 te grupe të caktuara etnike.
HYRJE
Alkaptonuria është sëmundje shumë e rrallë gjenetike që trashëgohet në mënyrë autosomale-recesive, dhe për këtë arsye shumë individë e kanë këtë sëmundje pa simptoma, duke mos qenë të vetëdijshëm për gjendjen e tyre deri në periudhën e moshës së pjekurisë. Pacientët nuk kanë nivele të mjaftueshme funksionale të enzimës së nevojshme për të degraduar acidin homogjenetik, duke çuar në shfaqjen e urinës së errët (të zezë). Alkaptonuria është ndër sëmundjet e para gjenetike që tregon se ndjekin parimet e trashëgimisë autosomale-recesive sipas Mendelit. Historikisht, besohet se mumja egjiptiane Harva është rasti i parë klinik i alkaptonurisë që daton mbi 1500 vjet para erës sonë. Sëmundja ndikon po aq te meshkujt dhe femrat, megjithatë simptomat kanë tendencë të shfaqen më herët te fëmijët meshkuj të lindur të gjallë. Rajonet me frekuencë më të lartë të kësaj sëmundjeje janë identifikuar në Sllovaki, Gjermani dhe Republikën Dominikane.
ETIOLOGJIA
Sëmundja zhvillohet si pasojë e mungesës së gjenit HGD, domethënë mungesës së enzimës homogjentiza oksidaza, e cila është e nevojshme për shpërbërjen e aminoacideve fenilalaninë dhe tirozinë. Kështu, pacientët që kanë këtë sëmundje janë homozigote ose heterozigote të ndërlikuara për mutacione që humbasin funksionin e këtij gjeni. Që një foshnje të zhvillojë alkaptonuri, është e nevojshme që të trashëgojë dy kopje të gjenit të defektuar HGD, ndërsa mundësitë janë shumë të vogla, prandaj kjo gjendje është vërtet e rrallë. Kur ka mungesë, acidi homogjentik grumbullohet në trup dhe në fund lirohet në urinën, duke i dhënë një ngjyrë të errët kur ekspozohet ndaj ajrit. Dëmtimi i indeve është pasojë e depozitimit të pigmentit që është i ngjashëm me melaninin (forma polimerike e BQA), i cili mund të shkaktojë shumë reaksione dhe prodhimin e radikaleve të lira, duke çuar kështu në dëmtim të mëtejshëm të indeve.
SIMPTOMAT DHE DIAGNOSTIKIMI
Një nga shenjat më të hershme të alkaptonurisë është ngjyrosja e errët e peleneve te foshnjat, prandaj kjo shenjë nuk duhet të injorohet ose të lihet pas dore që të sigurohet se çrregullimi nuk mbetet i pavërejtur. Përveç ndryshimit të ngjyrës së urinës në fëmijëri, pacientët zakonisht nuk zhvillojnë simptoma të tjera deri në moshën e rritur (zakonisht rreth të 20-tave deri në të 30-tat), e për këtë arsye nuk janë të vetëdijshëm për praninë e kësaj sëmundjeje deri më vonë. Gjatë jetës acid homogjentizik fillon të depozitohet dhe të grumbullohet në pjesë të ndryshme të trupit, duke shkaktuar simptoma karakteristike dhe dëmtime, siç janë:
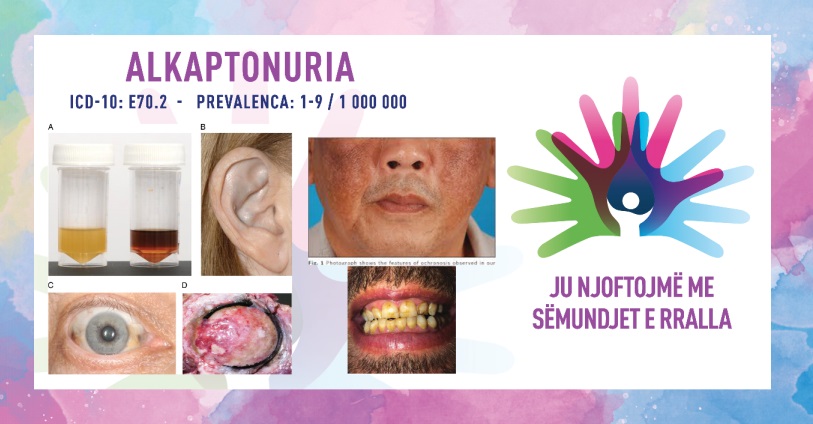
- ndryshimi i ngjyrës së urinës– simptomë më e zakonshme, urina bëhet e errët kur ekspozohet në ajër;
- ohronozë– ndryshim i ngjyrës së indit lidhës, duke përfshirë kockat dhe lëkurën; mund të ndikojë në zona të ndryshme, veçanërisht në veshë, hundë, sklerë dhe thonjtë;
- probleme me nyjet– shumë pacientë zhvillojnë artrit, veçanërisht në nyjet më të mëdha, siç janë supet, këmbët dhe gjunjët, për shkak të grumbullimit të pigmenteve në kocka;
- probleme me zemrën– depozitat mund të grumbullohen edhe në valvulat e zemrës, çka mund të çojë në probleme kardiovaskulare si stenozë, aritmi, dështim të zemrës etj.;
- gurë në veshka– nivelet e larta të acidit homogjentizik mund të shkaktojnë formimin e gurëve në veshka.
Pasi ekzistojnë disa diagnoza diferenciale (porfiria akute intermitente, artriti reumatoid, spondiliti ankiloz, osteoartriti), është e nevojshme të mblidhet një anamnezë e detajuar dhe të kryhen disa teste për të konfirmuar sëmundjen. Një mjet kyç diagnostikues është testi urinar, ku mostra bëhet e errët kur ekspozohet ndaj ajrit. Niveli i rritur i acidit homogjentizik mund të konfirmohet përmes analizës kimike. Po ashtu mund të bëhet testimi gjenetik për të identifikuar mutacionet në gjenin HGD, që do të sigurojë një diagnozë përfundimtare dhe më pas do të ndiqet këshillimi gjenetik. Incizimi me rreze X ose MRI mund të përdoret për të vlerësuar dëmtimin e nyjeve dhe për të ndjekur progresin e artritit.
PASOJAT
Jetëgjatësia e pacientëve që kanë alkaptonuri nuk është reduktuar ndjeshëm në krahasim me jetëgjatësinë mesatare, por dhimbja e vazhdueshme mund të çojë në rënie progresive të funksionit duke çuar në humbjen e lëvizshmërisë, kështu që pacientët shpesh përdorin mjete ndihmëse lëvizëse. Përveç kësaj, komplikimet kardiake shpesh janë të rrezikshme për jetën dhe mund të përkeqësojnë gjendjen.
TRAJTIMI
Edhe pse nuk ka trajtim që mund të shërojë sëmundjen, trajtimi fokusohet në lehtësimin e simptomave dhe përmirësimin e cilësisë së jetës. Terapia bazohet në: medikamente anti-inflamatore jo-steroide (AINS), të cilat përdoren më së shpeshti për të trajtuar dhimbjet në nyje dhe inflamacionin. Ndryshimet në dietë janë të dobishme, edhe pse nuk ka një dietë specifike për trajtimin e alkaptonurisë, disa rekomandime përfshijnë kufizimin e marrjes së proteinave, e kështu edhe të tirozinës dhe fenilalaninës për të kufizuar prodhimin e acidit homogjentizik. Terapia fizikale dhe operacionet përdoren në fazat më të avancuara, kur funksioni i nyjeve është i kufizuar, kur artriti është shumë i avancuar, kur ka humbje të lëvizshmërisë në nyje ose kur dëmtimi është aq i madh saqë kërkon ndërhyrje të duhur kirurgjikale. Hulumtimet e reja ofrojnë shpresë për lloje të reja terapie dhe shërim të mundshëm të sëmundjes. Megjithatë, ndërgjegjësimi për këtë gjendje mbetet thelbësor për diagnozën e hershme dhe kujdesin efikas.