Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
КОКЕЈНОВ СИНДРОМ/COCKAYNE SYNDROME – NEILL-DINGWALL SYNDROME
MKБ-10 Q87.1
ПРЕВАЛЕНЦА – непозната
ВОВЕД
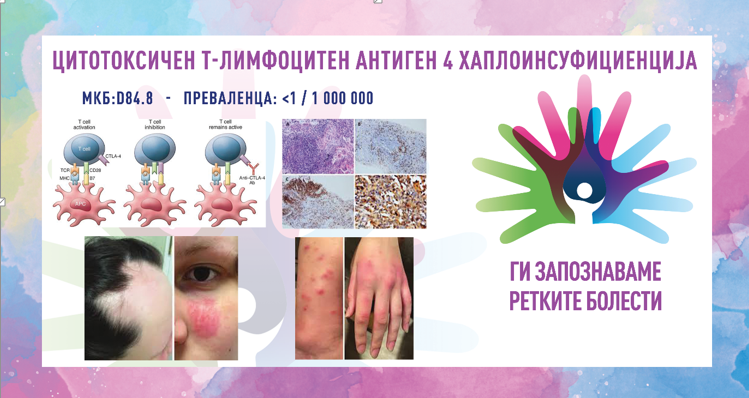
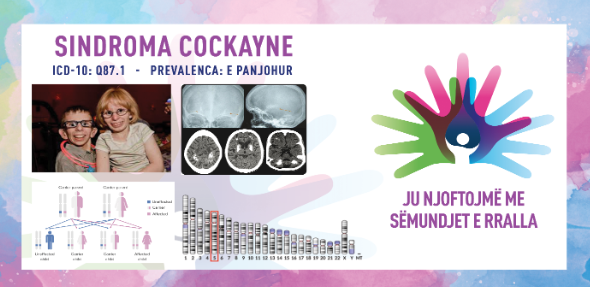
Кокејнов синдром е ретко заболување кое се карактеризира со абнормално мала големина на главата, неуспех за напредок во висина и тежина што доведува до џуџест раст и одложен развој. Заболените со ова нарушување покажуваат уникатни карактеристики на лицето, вклучително и вдлабнати очи, клунест нос и истакнати уши, а исто така доживуваат прогресивна деменција. Знаците и симптомите за оваа состојба обично се видливи уште од детството и тие се влошуваат со текот на времето. Кокејновиот синдром се дели на типови I, II и III врз основа на сериозноста и возраста од почетокот на симптомите.
ЕТИОЛОГИЈА
Синдромот е предизвикан од промени (патогени варијанти) во гените ERCC6 (CSA) и ERCC8 (CSB). Овие гени се вклучени во нормалната поправка на ДНК што се јавува по оштетување од ултравиолетовата светлина и сочинуваат околу 70% од случаите со генската мутација ERCC6 (CSA). Изложеноста на ултравиолетовата компонента на сончевата светлина, зрачењето и слободните радикали во телото ја оштетува ДНК и бидејќи клетките веќе не се во можност да ја поправат оштетената ДНК, таа се акумулира во клетките. Протеините направени од овие гени се вклучени во поправка на оштетената ДНК преку механизмот за поправка поврзана со транскрипција, особено ДНК во активните гени. Ако генот ERCC6 или ERCC8 е изменет (како кај Кокејнов синдром), оштетувањето на ДНК што се сретнува за време на транскрипцијата, не се поправа, предизвикувајќи РНК-полимеразата да биде позиционирана на таа локација, попречувајќи ја генската експресија. Како што се акумулира несанираното оштетување на ДНК, така е попречена прогресивно-поактивната генска експресија, што доведува до дефект на клетките или клеточна смрт, што веројатно придонесува за знаците на Кокејнов синдром, како што се предвремено стареење и невронска хипомиелинизација. Кокејновиот синдром се наследува автосомно рецесивно и ризикот е ист за машките и женските пациенти.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Карактеристични симптоми на Кокејнов синдром се:
нарушувања на растот и физичките карактеристики – прогресивното ограничување на растот се јавува на возраст од 1 до 2 години и се карактеризира со неуспех за напредок и микроцефалија; како што стареат децата, тие го губат поткожното масно ткиво, што резултира со прерано стареење на лицето и дисморфни карактеристики, како што се истакнати уши, вдлабнати очи, клунест нос и карактеристично тело со тенки екстремитети;
невролошки нарушувања и интелектуален развој – невролошките манифестации постепено напредуваат и произлегуваат од демиелинизација; засегнатите деца можат да изразат низа симптоми, вклучително и прогресивно сензоневрално губење на слухот што доведува до длабока глувост, интелектуални попречености, тешкотии при одење, нестабилно одење, лоша рамнотежа, епилепсија во некои случаи, напорен и неразбирлив говор и полиневропатии, притоа најчеста е сензомоторната демиелинизирачка полиневропатија;
наоди на кожата – тенка и чувствителна кожа, која пациентите ги прави подложни на тешки изгореници од сонце; cупкутаната липоатрофија придонесува за развој на вдлабнати очи и предвремено стареење на прогеричен изглед; вообичаените наоди вклучуваат цијанотични акрални едеми на екстремитетите, дистрофии на ноктите и аномалии на косата;
абнормалности на забите – мали, неправилни и ненормално распоредени, повеќе склони кон кариес отколку просечната популација;
мускулно-скелетни – спастичност на нозете, атаксија и контрактури во колковите, колената и глуждовите, издолжени екстремитети, преголеми шаки и стапала;
окуларни – често се јавуваат пигментирана ретинопатија, оптичка атрофија, миотични зеници, страбизам, катаракта, блефарокератоконјуктивитис и нистагмус;
ендокрината дисфункција – хипогонадизам кај мажите, кој се јавува во околу 30% од случаите и неправилна менструација кај жените како резултат на ендокрина дисфункција.
Дијагнозата се поставува врз основа на клиничката слика, клиничките карактеристики и резултатите од тестовите кои во себе вклучуваат:
генетско тестирање – со помош на примерок до крв се испитува постоењето на мутациите на генот ERCC6 или ERCC8;
биопсија на кожа;
МРИ на мозок – може да се открие евентуално постоење на дифузна хипомиелинизација на церебралната бела материја, калцификации во путаменот и вермиска атрофија.
ПОСЛЕДИЦИ
Последиците вклучуваат абнормално висок крвен притисок (хипертензија), бубрежна инсуфициенција, предвремена атеросклероза, гастроезофагеален рефлукс, прогресивна периферна моторна и сензорна невропатија што предизвикува потешкотии при одењето, интелектуална попреченост, доцнење во развојот, неуспех на растот, прогресивна пигментна ретинопатија, катаракта, сензоневрален губиток на слухот, зглобни контрактури, атаксија, тремор, напади, спастичност. Повеќето заболени имаат зголемена чувствителност на сончева светлина и сериозна реакција на антибиотик, наречен метронидазол, како и прерана смрт.
ТРЕТМАН
Децата со сомнение за овој синдром се подложни на мултидисциплинарна евалуација, вклучувајќи клинички преглед, лабораториски и имиџинг-студии, првенствено за да се исклучат другите дијагнози. Сеопфатната евалуација вклучува генетски испитувања, офталмолошки преглед со електроретинографија, невролошки со електроенцефалографија и студии за нервна спроводливост, како и гастроинтестинални, аудиолошки, дерматолошки, стоматолошки и ендокринолошки испитувања.
Третманот на Кокејнов синдром е симптоматски и поддржувачки. Пристапот може да вклучува специјално образование, физикална терапија и други медицински, социјални и/или стручни услуги. Се препорачува генетско советување за членовите на семејството. Децата со тип I обично одржуваат добри комуникациски вештини и покрај болеста. Се препорачува поттикнување на социјализација и образование соодветно на возраста. Од друга страна, децата со тип II се соочуваат со ограничени можности за интеракција со нивната околина поради сериозноста на сензорните и интелектуалните оштетувања, а смртта обично се случува во првата деценија. Спротивно на тоа, пациентите со тип III покажуваат релативно нормален ран развој, посетувајќи формално образование. Сепак, визуелните и аудитивните оштетувања, заедно со слабоста, се развиваат во адолесценцијата.
SINDROMA COCKAYNE / COCKAYNE SYNDROME – NEILL–DINGWALL SYNDROME
ICD-10 Q87.1
PREVALENCA – e panjohur
HYRJE
Sindroma Cockayne është sëmundje e rrallë që karakterizohet nga madhësi e vogël dhe jonormale e kokës, moszhvillim në gjatësi dhe peshë që çon në rritje xhuxhe dhe zhvillim të vonuar. Të prekurit nga ky çrregullim shfaqin veçori unike të fytyrës, duke përfshirë sy të zhytur, hundë të lakuar dhe veshë të spikatur, si dhe përjetojnë demencë progresive. Shenjat dhe simptomat zakonisht janë të dukshme që në fëmijëri dhe përkeqësohen me kalimin e kohës. Sindroma Cockayne ndahet në llojet I, II dhe III bazuar në ashpërsinë dhe moshën e fillimit të simptomave.
ETIOLOGJIA
Sindroma shkaktohet nga ndryshimet (variantet patogjenike) në gjenet ERCC6 (CSA) dhe ERCC8 (CSB). Këto gjene janë të përfshira në riparimin normal të ADN-së që ndodh pas dëmtimit nga drita ultravjollcë dhe përbëjnë rreth 70% të rasteve me mutacionin gjenetik ERCC6 (CSA). Ekspozimi ndaj përbërësit ultravjollcë të dritës së diellit, rrezatimit dhe radikaleve të lira në trup dëmton ADN-në, por pasi qelizat nuk janë më të afta të riparojnë ADN-në e dëmtuar, ajo grumbullohet në qeliza. Proteinat e krijuara nga këto gjene janë të përfshirë në riparimin e ADN-së së dëmtuar përmes mekanizmit të riparimit të lidhur me transkriptimin, veçanërisht ADN-së në gjenet aktive. Nëse gjeni ERCC6 ose ERCC8 është i ndryshuar (si në sindromën Cockayne), dëmtimi i ADN-së që ndodh gjatë transkriptimit nuk riparohet, duke bërë që polimeraza e ARN-së të pozicionohet në atë vend, duke penguar shprehjen e gjeneve. Ndërsa dëmtimi i pa riparuar i ADN-së grumbullohet, shprehja progresive aktive e gjeneve pengohet, gjë që çon në defekt të qelizave ose vdekje qelizore, e cila ka gjasa të kontribuojë në shenjat e sindromës Cockayne, si plakja e parakohshme dhe hipomielinizimi nervor. Sindroma Cockayne trashëgohet në mënyrë autosomale-recesive dhe rreziku është i njëjtë për pacientët meshkuj dhe femra.
SIMPTOMAT DHE DIAGNOSTIKIMI
Simptomat karakteristike të sindromës Cockayne janë:
çrregullimet e rritjes dhe karakteristikat fizike – kufizimi progresiv i rritjes shfaqet në moshën 1 deri në 2 vjeç dhe karakterizohet nga mungesa e përparimit në gjatësi dhe mikrocefalia; me kalimin e kohës, fëmijët humbin indin dhjamor nënlëkuror, duke rezultuar në plakje të hershme të fytyrës dhe karakteristika dismorfike, si veshë të spikatur, sy të zhytur, hundë të lakuar dhe një trup karakteristik me gjymtyrë të holla;
çrregullimet neurologjike dhe zhvillimi intelektual – manifestimet neurologjike përparojnë gradualisht dhe rrjedhin nga demielinizimi; fëmijët e prekur mund të shfaqin një sërë simptomash, duke përfshirë humbjen progresive të dëgjimit sensorineural që çon në shurdhim të thellë, aftësi të kufizuara intelektuale, vështirësi në ecje, ecje të paqëndrueshme, dobësi në ekuilibrim , epilepsi në disa raste, të folur me mundim dhe të pakuptueshëm, dhe polineuropati, ku më e zakonshmja është polineuropatia demielinizuese sensomotorike;
ndryshimet në lëkurë – lëkurë e hollë dhe e ndjeshme, e cila i bën pacientët të prirur për djegie të rënda nga dielli; lipoatrofia nënlëkurore kontribuon në zhvillimin e syve të zhytur dhe plakjen e hershme me një pamje progjerike; gjetjet e zakonshme përfshijnë edemë akrale cianotike në gjymtyrë, distrofi të thonjve dhe anomali të flokëve;
çrregullimet e dhëmbëve – dhëmbë të vegjël, të parregullt dhe të shpërndarë në mënyrë jonormale, më të prirur ndaj prishjes krahasuar me popullatën mesatare;
çrregullimet muskulo-skeletike – spasticitet i këmbëve, ataksi dhe kontraktura në ije, gjunjë dhe kyçe, gjymtyrë të zgjatura, duar dhe këmbë tepër të mëdha;
ndryshimet Okulare – shpesh shfaqen retinopatia pigmentare, atrofia optike, pupila miotike, strabizmi, katarakti, blefarokeratokonjuktiviti dhe nistagmus;
disfunksioni endokrin – hipogonadizëm te meshkujt, i cili shfaqet në rreth 30% të rasteve dhe menstruacione të parregullta te femrat si rezultat i dysfunksionit endokrin.
Diagnoza vendoset bazuar në tablonë klinike, karakteristikat klinike dhe rezultatet e testeve, të cilat përfshijnë:
testim gjenetik – përmes një kampioni gjaku testohet prania e mutacioneve të gjeneve ERCC6 ose ERCC8;
biopsi lëkure;
MRI e trurit – mund të zbulohet prania e mundshme e hipomielinizimit difuz të lëndës së bardhë cerebrale, kalcifikimeve në putamen dhe atrofi të vermisit.
PASOJAT
Pasojat përfshijnë presion të lartë të gjakut (hipertension), insuficiencë renale, aterosklerozë të parakohshme, refluks gastroezofageal, neuropati motorike dhe ndijore periferike progresive që shkakton vështirësi në ecje, aftësi të kufizuara intelektuale, vonesa në zhvillim, dështim në rritje, retinopati pigmentare progresive, katarakt, humbje dëgjimi sensorineural, kontraktura të nyjave, ataksi, tremor, kriza epileptike, spasticitet. Shumica e të prekurve kanë ndjeshmëri të shtuar ndaj rrezeve të diellit dhe reagim serioz ndaj antibiotikut të quajtur metronidazol, si dhe vdekje të parakohshme.
TRAJTIMI
Fëmijët me dyshimin për këtë sindromë i nënshtrohen një vlerësimi multidisiplinar, përfshirë ekzaminimin klinik, laboratorik dhe studimet e imazherisë, kryesisht për të përjashtuar diagnoza të tjera. Vlerësimi gjithëpërfshirës përfshin testime gjenetike, ekzaminim okulistik me elektroretinografi, neurologjik me elektroencefalografi dhe studime për përcjellshmërinë nervore, si dhe ekzaminime gastrointestinale, audiologjike, dermatologjike, stomatologjike dhe endokrinologjike. Trajtimi i sindromës Cockayne është simptomatik dhe mbështetës. Qasja mund të përfshijë arsimim të specializuar, terapi fizike dhe shërbime të tjera mjekësore, sociale dhe/ose profesionale. Këshillimi gjenetik rekomandohet për anëtarët e familjes. Fëmijët me tipin I zakonisht ruajnë aftësi të mira komunikimi pavarësisht nga sëmundja. Rekomandohet nxitja e socializimit dhe edukimit të përshtatshëm për moshën. Nga ana tjetër, fëmijët me tipin II përballen me mundësi të kufizuara për ndërveprim me mjedisin e tyre për shkak të ashpërsisë së dëmtimeve ndijore dhe intelektuale, ndërsa vdekja zakonisht ndodh në dekadën e parë të jetës. Në të kundërt, pacientët me tipin III tregojnë një zhvillim relativisht normal të hershëm, duke ndjekur arsimim formal. Megjithatë, dëmtimet vizuale dhe dëgjimore, së bashku me dobësinë, zhvillohen në adoleshencë.