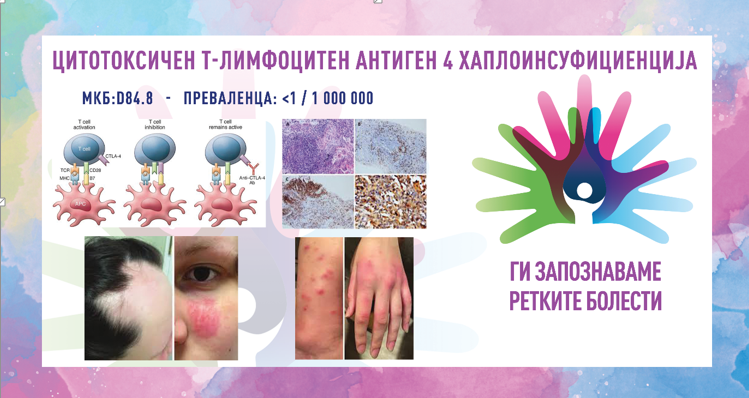
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
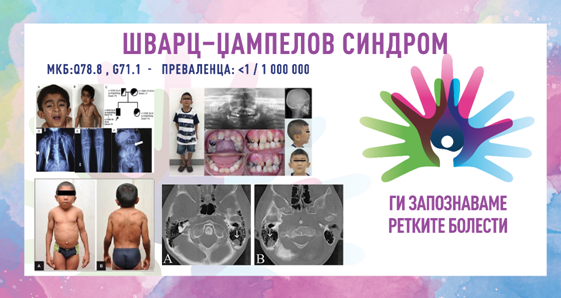
ШВАРЦ–ЏАМПЕЛОВ СИНДРОМ/ХОНДРОДИСТРОФИЧНА МИОТОНИЈА/ SCHWARTZ–JAMPEL SYNDROME (SJS)/CHONDRODYSTROPHIC MYOTONIAMKБ-10 Q78.8, G71.1
ПРЕВАЛЕНЦА – забележана кај помалку од 1/100 000 население (во литературата до денес се опишани 130 случаи).
Шварц–Џампеловиот синдром (SJS) или хондродистрофична миотонија е ретка генетска болест предизвикана од мутација на генот на перлекан (HSPG2), што предизвикува остеохондродисплазија поврзана со миотонија. Тоа е генетски условен синдром кој ги
вклучува мускулите и нервниот систем, и кој води до значителни физички и невролошки нарушувања, особено во поглед на мускулниот тонус и контролата на движењата. Типичните карактеристики на болеста вклучуваат ниска висина, абнормалности на скелетот, постојани мускулни контракции и хипертрофија на мускулатурата. Синдромот првпат е опишан во 1962 година од американскиот офталмолог Оскар Шварц и американскиот невроофталмолог Роберт Стивен Џампел. Поради својата реткост, оваа болест не е широко позната, и иако пациентите имаат речиси нормален животен век, сепак синдромот има сериозно влијание врз квалитетот на животот на пациентите
ЕТИОЛОГИЈА
Шварц–Џампеловиот синдром е предизвикан од мутации во генот HSPG2, кој го кодира протеинот перлекан позициониран во мускулите и рскавицата. Перлеканот е важен за᾿ функцијата на мускулите и нервите бидејќи ги регулира јонските канали кои се вклучени во преносот на нервните импулси. Кај синдромот постои сомневање дека абнормалната функција на перлекан доведува до недостаток на ацетилхолинестераза, ензим вклучен во разградувањето на невротрансмитерот ацетилхолин, кој ја поттикнува мускулната контракција. Ако ацетилхолинот не се разложи, може да доведе до продолжена мускулна контракција/зацврстување на мускулите (миотонија). Шварц–Џампеловиот синдром се наследува автосомно рецесивно, што значи дека се потребни две копии од мутираниот ген (една од секој родител) за да се развие болеста, иако некои пријавени случаи укажуваат на автосомно доминантна шема на наследување. Родителите на детето со автосомно рецесивна состојба обично не боледуваат од таа состојба. Генетскиот локус е мапиран на 1p36.1 Некои деца од опишаните случаи во литературата имале благи скелетни промени (SJS-1), додека други имале примарна коскена дисплазија придружена со грчеви, односно миотонија (SJS-2). Првата група покажала поврзаност со хромозом 1p36.1-p34, додека втората група не покажува таква поврзаност.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Симптомите на Шварц–Џампеловиот синдром вклучуваат широк спектар на физички и невролошки проблеми. Главните симптоми се
1. миотонија – тешкотија при опуштање на мускулите по нивната контракција; се манифестира со грчење на мускулите по изведување на движења, изразито се влошува при ниска надворешна температура или при стрес;
2. мускулни спазми, покачен мускулен тонус – предизвикува затегнатост на мускулите и ограничувања во движењето, како и појава на хипертрофија на мускулите на телото;
3. прогресивна слабост – најчесто се јавува неподносливост на физичка активност и слабост на мускулите, особено на екстремитетите;
4. очи – овој синдром може да биде поврзан со катаракта, блефароспазам (спазам на капакот) и други визуелни нарушувања;
5. кардиоваскуларни проблеми – иако не се толку чести, сепак можат да се појават, како на пример, абнормалности во работата на срцето;
6. коскени и зглобни деформитети – краток врат, конвексен граден кош, абнормална кривина на пршлените, криво стапало и/или рамни стапала, деформитет на колкот што вклучува зголемен агол меѓу вратот и телото на бутната коска, неправилност на капителарните епифизи на бутната коска, хондродисплазија, остеопороза
SJS-1 – соодветствува на оригиналниот опис на Шварц и Џампел. Коскените деформитети не се истакнати при раѓање, миотонијата на лицето е главната карактеристика која создава специфична тријада што вклучува стеснување на очните прорези (блефарофимоза/блефароспазам), собирање на устата и брчкање на брадата. Стимулацијата, како на пример, лесен удар или дување врз очните капаци, предизвикува блефароспазам. Континуираната миотонија на екстремитетите предизвикува вкочанетост при одење и нетолеранција на физичка активност. Иако, моторниот развој за време на првата година е бавен, сепак интелигенцијата е нормална.
SJS-2 – се карактеризира со изразени коскени деформитети при раѓање, кои сугерираат на Моркиов синдром (остеохондродистрофија). Неонаталната смртност кај оваа форма е висока.Шварц–Џампеловиот синдром се дијагностицира врз основа на карактеристичните црти на лицето, скелетните карактеристики и миотонијата. Тестовите на крвта можат да покажат покачена серумска креатин киназа или алдолаза. Рендгенските зраци, мускулната биопсија или електромиографијата (ЕМГ) можат да бидат од помош за засегнатите лица. Електромиографијата (ЕМГ) е специфична и клучна при поставувањето на дијагнозата и покажува континуирана активност на моторните единици. Првенствено, оваа активност се интерпретирала како миотонија. Миотонија може да биде присутна, но континуираната активност на моторните единици е одговорна за симптомите на лицето и екстремитетите. Серумската концентрација на креатин киназа (CK) може да биде благо зголемена. Хистолошкиот изглед на мускулот обично е нормален, но може да покаже варијации во големината на мускулните влакна и зголемен број на централни јадра. Генетското тестирање за генот HSPG2 може да ја потврди дијагнозата.
ПОСЛЕДИЦИ
Доколку болеста се стабилизира по адолесценцијата, тогаш истата не влијае на нормалниот животен век. Сепак, Шварц–Џампеловиот синдром има сериозни последици за пациентите и тие обично вклучуваат хронични и прогресивни невролошки и мускулни проблеми
– хронична мускулна слабост – пациентите често имаат тешкотии во движењето, што може да доведе до потешкотии во извршувањето на секојдневни активности; мускулната слабост може да биде прогресивна, што значи дека со текот на времето ќе се зголемува потребата од помош при извршување на обичните задачи;
– проблеми со видот – честа појава се катаракта, блефароспазам (неволен спазам на капакот) и други визуелни нарушувања, и тие можат значително да го нарушат видот и квалитетот на животот на пациентите;
– проблеми со функцијата на срцето – кај некои пациенти можат да се појават кардиоваскуларни компликации, како што се абнормалности во срцевиот ритам или срцевите дефекти;
– психолошки ефекти – постојаната мускулна болка, ограниченото движење и хроничниот замор можат да доведат до депресија, анксиозност и други психолошки нарушувања, со што дополнително се влошува состојбата на пациентот;
– влијание на животен век – самата болест има тенденција да се стабилизира во адолесцентниот период и не влијае врз животниот век, но во ретки случаи, тешките последици од Шварц–Џампеловиот синдром можат да доведат до скратен животен век, особено ако сериозните компликации не се третираат навремено.
ТРЕТМАН
Третманот на Шварц–Џампеловиот синдром во голема мера е насочен кон ублажување на симптомите и подобрување на квалитетот на животот на пациентите. Пошироко, третманот вклучува комбинација на лекови, физиотерапија и хируршка интервенција во зависност од сериозноста на симптомите.
Главните пристапи за третман вклучуваат:
1. лекови за миотонија – лекови, како што се дијазепам или други антиконвулзивни лекови, често се користат за намалување на миотонијата (тешкотијата при опуштање на мускулите по нивната контракција); овие лекови помагаат во олеснување на грчевите и ја намалуваат мускулната затегнатост;
2. физиотерапија и рехабилитација – за да се спречи влошување на мускулната слабост и да се подобри мобилноста, пациентите обично следат програма за
физиотерапија; физиотерапевтите користат различни техники за вежби и мобилизации на мускулите и зглобовите, што помага во одржување на нивната функција;
3. третман на очни нарушувања – може да се јави присуство на катаракта и на други проблеми со видот; катарактата може да се отстрани со хируршки третман, што може значително да го подобри видот;
4. психолошка поддршка – поради психолошките ефекти кои произлегуваат од хроничната болка и ограничената мобилност, пациентите имаат потреба од терапија за поддршка при нападите на депресија и анксиозност, како што се когнитивно-бихејвиоралната терапија и советувањето;
5. генетско советување – поради генетската природа на синдромот, генетското советување е од помош за семејствата кои можат да бидат засегнати од болеста; тоа им помага на родителите да ги разберат шансите за пренесување на болеста на следните генерации и да донесат паметни одлуки;
6. контрола на други компликации – за да се спречат или контролираат компликациите, како што се проблеми со срцето или дијабетес, пациентите можат да бидат лекувани со специјализирани лекови и редовни контроли.
Шварц–Џампеловиот синдром е сериозна, прогресивна болест која има големо влијание на физичкото и психолошкото здравје на пациентите. Иако не постои лек, раното откривање, соодветната терапија и мултидисциплинарниот пристап за третман можат да им помогнат на пациентите да го контролираат напредувањето на болеста и да го подобрат квалитетот на животот. За пациентите со Шварц–Џампеловиот синдром, интегрираниот третман и поддршката од страна на медицинскиот тим се клучни за постигнување добри резултати.
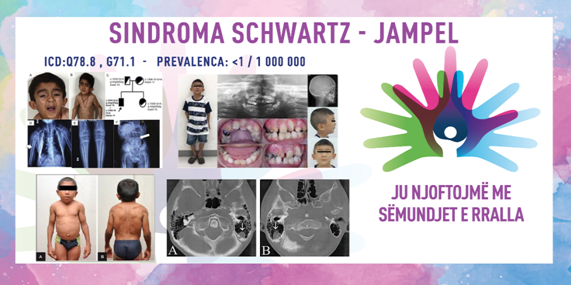
SINDROMA SCHWARTZ – JAMPEL/ MIOTONIA KONDRODISTROFIKE/ SCHWARTZ–JAMPEL SYNDROME (SJS)/ CHONDRODYSTROPHIC MYOTONIA
ICD-10 Q78.8, G71.1
PREVALENCA – vërehet te më pak se 1 në 100,000 banorë (deri më tani janë raportuar 130 raste në literaturë).
HYRJE
Sindromi Schwartz–Jampel (SJS) ose miotonia kondrodistrofike është sëmundje gjenetike e rrallë e shkaktuar nga një mutacion në gjene të perlekanit (HSPG2), që çon në osteokondrodisplazi të lidhur me miotoni. Ky është sindromë e trashëguar gjenetik që përfshin muskujt dhe sistemin nervor, duke çuar në çrregullime të rëndësishme fizike dhe neurologjike, veçanërisht në lidhje me tonusin muskulor dhe kontrollin e lëvizjeve. Karakteristikat tipike të sëmundjes përfshijnë shtat të shkurtër, abnormalitete skeletore, kontraktime të vazhdueshme muskulore dhe hipertrofi të muskujve. Sindromi u përshkrua për herë të parë në vitin 1962 nga oftalmologu amerikan Oscar Schwartz dhe neuroftalmologu amerikan Robert Steven Jampel. Për shkak të rrallësisë së saj, kjo sëmundje nuk është shumë e njohur, dhe ndonëse pacientët kanë një jetë pothuajse normale, sindromi ka një ndikim serioz në cilësinë e jetës së pacientëve.
ETIOLOGJIA
Sindromi Schwartz–Jampel shkaktohet nga mutacione në genin HSPG2, që kodon proteinën perlekan, e cila është e pranishme në muskuj dhe kartilagje. Perlekan është e rëndësishme për funksionimin e muskujve dhe nervave, pasi rregullon kanalet jonike që janë të përfshira në transmetimin e impulseve nervore. Te ky sindrom dyshohet që funksioni abnorm i perlekanit çon në mungesë të enzimës acetilkolinesterazë, një enzimë që është e përfshirë në çaktivizimin e neurotransmiterit acetilkolin, i cili shkakton kontraktimin e muskujve. Nëse acetilkolina nuk çaktivizohet, mund të shkaktojë kontraktim të vazhdueshëm të muskujve (miotoni). Sindromi i Schwartz–Jampel trashëgohet në mënyrë autosomale recesive, që do të thotë se duhen dy kopje të gjenit të modifikuar (një nga çdo prind) për të zhvilluar sëmundjen, megjithatë disa raste të raportuara tregojnë një model autosomal dominant të trashëgimisë. Prindërit e një fëmije me një gjendje autosomale recesive zakonisht nuk vuajnë nga kjo sëmundje. Lokusi gjenetik është i vendosur në 1p36.1. Disa fëmijë nga rastet e përshkruara në literaturë kanë pasur ndryshime të lehta skeletore (SJS-1), ndërsa të tjerë kanë pasur displazi primare të kockave të shoqëruara me krampe, përkatësisht miotoni (SJS-2). Grupi i parë ka treguar lidhje me kromozomin 1p36.1-p34, ndërsa grupi i dytë nuk ka treguar një lidhje të tillë.
SIMPTOMAT DHE DIAGNOSTIKIMI
Simptomat e Sindromit Schwartz–Jampel përfshijnë një gamë të gjerë të problemeve fizike dhe neurologjike. Simptomat kryesore janë:
1. miotoni – vështirësi në relaksimin e muskujve pas kontraktimit të tyre; manifestohen me krampe të muskujve pas lëvizjeve, dhe përkeqësohen në temperatura të ulëta ose gjatë stresit;
2. spazma muskulore, tonus i lartë muskulor – shkakton tension në muskuj dhe kufizime në lëvizje, si dhe shfaqje të hipertrofisë muskulore;
3. dobësi progresive – zakonisht vërehet te pacientët që nuk mund të kryejnë aktivitet fizik dhe përjetojnë dobësi muskulore, veçanërisht në ekstremitete;
4. probleme me sytë – ky sindrom mund të lidhet me kataraktën, spazmën e qepallës (blefarospazëm) dhe probleme të tjera vizuale;
5. probleme kardiovaskulare – megjithëse nuk janë të zakonshme, mund të shfaqen si abnormalitete në ritmin e zemrës;
6. deformitete kockore dhe artikulare – qafë e shkurtër, gjoks i konveksuar, kënd abnormal të vertebrave, shputa të shtrembëra ose të rrafshta, dhe deformitete të tjera kockore dhe artikulare.
SJS-1 – korrespondon me përshkrimin origjinal të Schwartz dhe Jampel. Deformimet kockore nuk janë të dukshme në lindje, miotonia e fytyrës është tipari kryesor që prodhon një treshe specifike që përfshin ngushtimin e të çarave të syrit (blefarofimoza/blefarospazma), rrudhat e gojës dhe rrudhat e mjekrës. Stimulimi, si një goditje e lehtë ose goditje në qepalla, shkakton blefarospazëm. Miotonia e vazhdueshme e gjymtyrëve shkakton ngurtësi gjatë ecjes dhe intolerancë ndaj aktivitetit fizik. Megjithëse zhvillimi motorik gjatë vitit të parë është i ngadalshëm, inteligjenca është normale.
SJS-2 – karakterizohet nga deformitete të theksuara kockore në lindje, që sugjerojnë sindromin Morkio (osteo-kondrodistrofi). Vdekshmëria neonatale është më e lartë në këtë formë.
Sindroma Schwartz-Jampel diagnostikohet në bazë të tipareve karakteristike të fytyrës, tipareve skeletore dhe miotonisë. Testet e gjakut mund të tregojnë rritje të kreatinë kinazës ose aldolazës në serum. Rrezet X, biopsia e muskujve ose elektromiografia (EMG) mund të jenë të dobishme për individët e prekur. Elektromiografia (EMG) është specifike dhe vendimtare në vendosjen e diagnozës dhe tregon aktivitet të vazhdueshëm të njësive motorike. Kryesisht, ky aktivitet u interpretua si miotonia. Miotonia mund të jetë e pranishme, por aktiviteti i vazhdueshëm i njësive motorike është përgjegjës për simptomat e fytyrës dhe gjymtyrëve. Përqendrimi i kreatinë kinazës (CK) në serum mund të jetë pak i ngritur. Pamja histologjike e muskujve është zakonisht normale, por mund të tregojë ndryshime në madhësinë e fibrave muskulore dhe një numër të shtuar të bërthamave qendrore. Testimi gjenetik për gjenin HSPG2 mund të konfirmojë diagnozën.
PASOJAT
Nëse sëmundja stabilizohet pas adoleshencës, atëherë nuk ndikon në jetëgjatësinë normale. Megjithatë, Sindromi Schwartz–Jampel ka pasoja serioze për pacientët dhe përfshin shpesh çrregullime kronike dhe progresive të muskujve dhe nervave.
dobësi kronike e muskujve – pacientët shpesh kanë vështirësi në lëvizje, gjë që mund të çojë në vështirësi në kryerjen e aktiviteteve të përditshme; dobësia e muskujve mund të jetë progresive, që do të thotë se me kalimin e kohës do të rritet nevoja për ndihmë në kryerjen e detyrave të zakonshme;
problemet e shikimit – kataraktet, blefarospazma (spazma e pavullnetshme e kapakut) dhe çrregullime të tjera të shikimit janë të zakonshme, të cilat mund të dëmtojnë ndjeshëm shikimin dhe cilësinë e jetës së pacientëve;
probleme me funksionin e zemrës – te disa pacientë mund të shfaqen komplikime kardiovaskulare, si anomalitë e ritmit të zemrës ose defekte të zemrës;
efektet psikologjike – dhimbje të vazhdueshme të muskujve, lëvizje të kufizuar dhe lodhje kronike mund të çojnë në depresion, ankth dhe çrregullime të tjera psikologjike, të cilat përkeqësojnë më tej gjendjen e pacientit;
ndikimi në jetëgjatësinë – vetë sëmundja tenton të stabilizohet në periudhën e adoleshencës dhe nuk ndikon në jetëgjatësinë, por në raste të rralla, pasojat e rënda të sindromës Schwartz-Jampel mund të çojnë në një jetëgjatësi të shkurtuar, veçanërisht nëse nuk ka komplikime serioze. trajtohen në kohë.
TRAJTIMI
Trajtimi i Sindromit Schwartz–Jampel ka për qëllim kryesisht lehtësimin e simptomave dhe përmirësimin e cilësisë së jetës së pacientëve. Më gjerësisht, trajtimi përfshin një kombinim të barnave, fizioterapisë dhe kirurgjisë, në varësi të ashpërsisë së simptomave. Metodat kryesore të trajtimit përfshijnë:
1. barnat për miotoninë – barnat, si diazepami ose barna të tjera antikonvulsante, përdoren shpesh për të reduktuar miotoninë (vështirësi në relaksimin e muskujve pasi ato tkurren); këto barna ndihmojnë në lehtësimin e spazmave dhe zvogëlojnë tensionin e muskujve;
2. fizioterapi dhe rehabilitim – për të parandaluar përkeqësimin e dobësisë muskulare dhe për të përmirësuar lëvizshmërinë, pacientët zakonisht ndjekin një program fizioterapie; fizioterapistët përdorin teknika të ndryshme për ushtrime dhe mobilizime të muskujve dhe kyçeve, gjë që ndihmon në ruajtjen e funksionit të tyre;
3. trajtimi i çrregullimeve të syrit – mund të ndodhë prania e kataraktave dhe problemeve të tjera të shikimit; kataraktet mund të hiqen me trajtim kirurgjik, i cili mund të përmirësojë ndjeshëm shikimin;
4. mbështetja psikologjike – për shkak të efekteve psikologjike që vijnë nga dhimbjet kronike dhe lëvizshmëria e kufizuar, pacientët kanë nevojë për terapi për të parandaluar sulmet e depresionit dhe ankthit, si terapia kognitive (e sjelljes) dhe këshillimi;
5. këshillimi gjenetik – për shkak të natyrës gjenetike të sindromës, këshillimi gjenetik është i dobishëm për familjet që mund të preken nga sëmundja; ai i ndihmon prindërit të kuptojnë shanset për ta përcjellë sëmundjen te brezat e ardhshëm dhe të marrin vendime të zgjuara;
6. kontrolli i komplikimeve të tjera – për të parandaluar ose kontrolluar komplikime, si problemet e zemrës ose diabeti, pacientët mund të trajtohen me barna të specializuara dhe kontrolle të rregullta.
Sindromi Schwartz-Jampel është sëmundje serioze, progressive, që ka një ndikim të madh në shëndetin fizik dhe psikologjik të pacientëve. Megjithëse nuk ka kurë, zbulimi i hershëm, terapia e përshtatshme dhe një qasje multidisiplinare ndaj trajtimit mund t’i ndihmojnë pacientët të kontrollojnë përparimin e sëmundjes dhe të përmirësojnë cilësinë e jetës. Për pacientët me sindromën Schwartz-Jampel, trajtimi i integruar dhe mbështetja nga ekipi mjekësor janë çelësi për arritjen e rezultateve të mira.