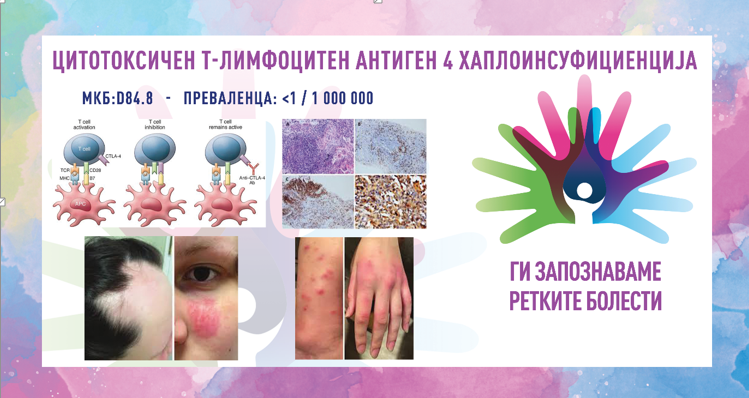
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
АКТИВИРАН ФОСФОИНОЗИТИД 3-КИНАЗА ДЕЛТА СИНДРОМ/
ACTIVATED PI3K DELTA SYNDROME (APDS)
MKБ-10 D81.82
ПРЕВАЛЕНЦА – овој синдром претставува ретка примарна имунодефициенција, со само 256 пријавени случаи на глобално ниво, погодувајќи ги подеднакво и двата пола.
ВОВЕД
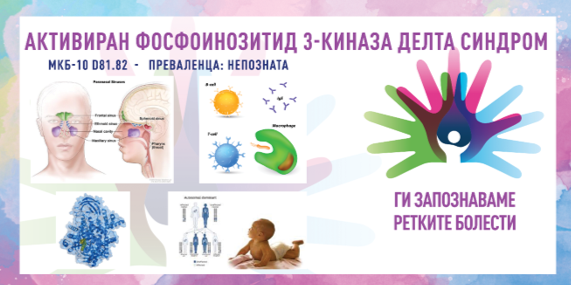
Синдромот на активиран фосфоинозитид 3-киназа делта (APDS) е ретка, наследна состојба која влијае на начинот на кој функционира имунолошкиот систем. Постојат два типа APDS: APDS1 се должи на промени во генот PIK3CD и APDS2 се должи на промени во генот PIK3R1. И двата типа имаат слични симптоми и се наследени како автосомно-доминантен модел во семејствата. Синдромот АФИ 3-КД првпат бил опишан во 2013 година и претставува еден од примарните имунодефициенции, при кои луѓето се раѓаат со имунолошки систем кој не функционира правилно и добиваат чести инфекции кои тешко се лекуваат.
ЕТИОЛОГИЈА
Синдромот АФИ 3-КД е предизвикан од патогени варијанти во генот PIK3CD (APDS1) или во генот PIK3R1 (APDS2). Токму тие гени се важни за растот, опстанокот и функцијата на одредени типови имунолошки клетки, наречени Т-клетки и Б-клетки. Тие претставуваат специјализирани бели крвни зрнца кои помагаат во борбата против инфекциите. Патогените варијанти на овие два гена влијаат на тоа како растат и функционираат Б-клетките и Т-клетките. Луѓето со синдромот АФИ 3-КД често имаат Б-клетки и Т-клетки кои не функционираат нормално, што доведува до инфекции, воспаление на цревата и автоимуна болест. APDS1 и APDS2 се наследуваат во доминантна шема во семејствата. Доминантните генетски нарушувања се јавуваат кога е неопходна само една копија од мутираниот ген за да предизвика одредена болест. Мутираниот ген може да биде наследен од кој било родител или може да биде резултат на променет ген кај засегнатата индивидуа. Ризикот од пренесување на мутираниот ген од засегнат родител на потомство е 50 % за секоја бременост. Ризикот е ист за машките и женските лица.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Синдромот АФИ 3-КД влијае различно на секоја личност. Некои луѓе имаат многу благи симптоми, додека други се многу посериозно погодени. Повеќето луѓе со синдромот АФИ 3-КД ја добиваат првата инфекција во раното детство, но симптомите можат да започнат на која било возраст. Најчестите симптоми на синдромот АФИ 3-КД се чести инфекции на горниот респираторен тракт, инфекции на синусите, инфекции на увото, бронхитис и пневмонија (инфекција на белите дробови). Другите карактеристични симптоми вклучуваат иритација на гастроинтестиналниот тракт, отекување на лимфните јазли, зголемен црн дроб и слезина, и зголемен ризик за лимфом. Со текот на времето, честите инфекции на увото и респираторниот тракт можат да доведат до трајно губење на слухот и лузни на белите дробови (бронхиектазии). Ова е почесто кај APDS1 отколку кај APDS2. Исто така, чести се и вирусните инфекции, особено инфекциите со вирусот Епштајн-Бар и вирусите на херпес симплекс. Овие инфекции можат да бидат тешки за лекување и можеби никогаш целосно нема да исчезнат. Луѓето со синдромот АФИ 3-КД можат да развијат зголемени лимфни јазли, како и зголемен црн дроб (хепатомегалија) или слезина (спленомегалија). Може да се појави и воспаление на цревата што доведува до тешка дијареја која понекогаш резултира со хоспитализација. Инфекциите на кожата исто така се чести. Кај луѓе со синдромот АФИ 3-КД исто така се забележани задоцнување на растот, низок раст и доцнење во невроразвојот. Понискиот раст од просечниот е почест кај APDS2. Луѓето со овој синдром исто така имаат зголемен ризик од развој на лимфом и други видови рак на крвта.
Синдромот АФИ 3-КД се дијагностицира врз основа на симптомите, клинички преглед, детална семејна историја и лабораториско тестирање со цел да се најдат некои абнормалности во нивоата и функцијата на имуните клетки. Бидејќи синдромот АФИ 3-КД може да наликува на друга имунодефициенција, неопходно е генетско тестирање за да се постави специфична дијагноза. Бидејќи е наследна, членовите на семејството на потврден пациент треба да бидат генетски тестирани и покрај тоа што можеби ги немаат истите симптоми или немаат никакви симптоми, сепак можат да ја носат генетската варијанта и да ја пренесат на своите деца.
ПОСЛЕДИЦИ
Прилагодувањата на животниот стил потребни за лицата со синдромот АФИ 3-КД зависат од сериозноста на индивидуалните симптоми што ги има секое лице. Изложеноста на потенцијални бактерии и вируси кои би можеле да предизвикаат инфекција, може да се намали со практикување на добра лична хигиена, храна и животна средина; преземање мерки на претпазливост при пливање (избегнување езера/бара), минимизирање на контакт со нечистотија, земја или животни; и избегнување контакт со луѓе со инфекции.
Здравјето на пациентите треба да се следи редовно, вклучувајќи проверки за отечени лимфни јазли и почеток на знаци и симптоми на лимфом кои се типични, и тоа: висока температура, ноќно потење, замор, чешање и необјаснето губење на тежината.
ТРЕТМАН
Третманот за синдромот АФИ 3-КД е фокусиран на управување со симптомите, спречување инфекции и намалување на воспалението. Синдромот се третира со користење на комбинација на долготрајна заместителна терапија со имуноглобулин за да се помогне во поддршката на имунолошкиот систем и имуносупресивни лекови за да се помогне при симптомите поради автоимунитет и воспаление. Покрај тоа, на луѓето со синдромот АФИ 3-КД често им се потребни дневни антибиотици за да помогнат во спречувањето на инфекциите пред тие да се случат. Децата со синдромот АФИ 3-КД можат да имаат потреба од ушни цевки (цевки тимпаностомија) поради чести ушни инфекции. На некои можат да им се отстранат крајниците поради оток и инфекции на синусите. На луѓето со потежок степен на синдромот АФИ 3-КД може да им биде потребна респираторна поддршка, вклучувајќи дополнителен кислород и физиотерапија на градниот кош. Дополнително, физичката, професионалната и/или говорната терапија може да помогне при некои од долгорочните компликации. Трансплантација на хематопоетски матични клетки (HSCT) се користи за лекување на некои луѓе со синдромот АФИ 3-КД кои не реагираат на стандардна терапија. Хематопоетските матични клетки се развиваат во коскената срцевина и имаат потенцијал да станат каков било тип крвни клетки. HSCT вклучува замена на коскената срцевина со нови матични клетки кои можат да бидат здрави, функционални имунолошки клетки. HSCT се користи како третман за неколку примарни имунодефициенции, како и за одредени видови рак на крвта.

SINDROMA E KINEAZËS 3 TE FOSFOINOZIDIT TE AKTIVIZUAR DELTA/
ACTIVATED PI3K DELTA SYNDROME (APDS)
ICD-10 D81.82
PREVALENCA – kjo sindromë është deficencë imunitare primare e rrallë, me vetëm 256 raste të raportuara në nivel global, që ndikon në të dyja gjinitë në mënyrë të barabartë.
HYRJE
Sindroma e kinazës 3 të fosfoinozidit të aktivizuar delta (APDS) është gjendje e rrallë, trashëgimore që ndikon në mënyrën se si funksionon sistemi imunitar. Ekzistojnë dy lloje të APDS: APDS1, që shkaktohet nga ndryshimet në gjenin PIK3CD dhe APDS2, që shkaktohet nga ndryshimet në gjenin PIK3R1. Të dy llojet kanë simptoma të ngjashme dhe trashëgohen sipas një modeli autosomal-dominant në familje. Sindroma e APDS 3-KD është përshkruar për herë të parë në vitin 2013 dhe është nga deficencat imunitare primare, ku njerëzit lindin me një sistem imunitar që nuk funksionon siç duhet dhe përjetojnë infeksione të shpeshta që janë të vështira për t’u trajtuar.
ETIOLOGJIA
Sindroma e APDS 3-KD shkaktohet nga variacione patogjene në gjenin PIK3CD (APDS1) ose në gjenin PIK3R1 (APDS2). Këta gjene janë të rëndësishëm për rritjen, mbijetesën dhe funksionin e disa llojeve të qelizave imunitare, të quajtura qeliza T dhe qeliza B. Ato janë qeliza të specializuara të gjakut të bardhë që ndihmojnë në luftën kundër infeksioneve. Variacionet patogjene të këtyre dy gjeneve ndikojnë në mënyrën se si rriten dhe funksionojnë qelizat B dhe T. Njerëzit me sindromën APDS 3-KD shpesh kanë qeliza B dhe T që nuk funksionojnë normalisht, çka çon në infeksione, inflamacion të zorrëve dhe sëmundje autoimune. APDS1 dhe APDS2 trashëgohen në një model dominues në familje. Çrregullimet gjenetike dominante ndodhin kur është e nevojshme vetëm një kopje e gjenit të mutuar për të shkaktuar një sëmundje të caktuar. Gjeni i mutuar mund të trashëgohet nga cilido prind ose mund të jetë rezultat i një ndryshimi gjenetik te individi i prekur. Rreziku i transmetimit të gjenit të mutuar nga një prind i prekur te pasardhësit është 50% për çdo shtatzëni. Ky rrezik është i njëjtë për meshkujt dhe femrat.
SIMPTOMAT DHE DIAGNOSTIKIMI
Sindroma APDS 3-KD ndikon ndryshe te çdo individ. Disa njerëz kanë simptoma shumë të lehta, ndërsa të tjerë janë prekur shumë më rëndë. Shumica e njerëzve me sindromën APDS 3-KD e kanë infeksionin e parë në fëmijërinë e hershme, por simptomat mund të fillojnë në çdo moshë. Simptomat më të shpeshta të sindromës APDS 3-KD janë infeksionet e shpeshta të traktit respirator të sipërm, infeksionet e sinuseve, infeksionet e veshit, bronkiti dhe pneumonia (infeksioni i mushkërive). Simptoma të tjera karakteristike përfshijnë irritimin e traktit gastrointestinal, ënjtje të nyjeve limfatike, mëlçinë dhe shpretkë e zgjeruar, dhe rrezik të shtuar për limfomë. Me kalimin e kohës, infeksionet e shpeshta të veshit dhe të traktit respirator mund të çojnë në humbje të përhershme të dëgjimit dhe plagë të mushkërive (bronhiektazi). Kjo është më e shpeshtë te APDS1 sesa te APDS2. Gjithashtu, infeksionet virale janë të shpeshta, veçanërisht infeksionet me virusin Epstein-Barr dhe viruset e herpes simpleks. Këto infeksione mund të jenë të vështira për t’u trajtuar dhe mund të mos zhduken kurrë plotësisht. Njerëzit me sindromën APDS 3-KD mund të zhvillojnë nyje limfatike të zgjeruara, si dhe mëlçi të zgjeruar (hepatomegali) ose shpretkë(splenomegali). Mund të shfaqet edhe inflamacion i traktit të zorrëve që çon në diarre të rëndë, e cila ndonjëherë rezulton me shtrim në spital. Infeksionet e lëkurës janë gjithashtu të shpeshta. Te njerëzit me sindromën APDS 3-KD është vërejtur gjithashtu vonesë në rritje, rritje e ulët dhe vonesë në zhvillimin neurologjik. Rritja më e ulët se norma është më e shpeshtë te APDS2. Njerëzit me këtë sindromë gjithashtu kanë rrezik të shtuar për zhvillimin e limfomës dhe lloje të tjera të kancerit të gjakut.
Sindroma APDS 3-KD diagnostikohet mbi bazën e simptomave, ekzaminimit klinik, historisë të detajuar familjare dhe testimeve laboratorike për të gjetur abnormalitete në nivelet dhe funksionin e qelizave imunitare. Duke qenë se sindroma APDS 3-KD mund të ngjajë me një deficencë tjetër imunitare, është e nevojshme të kryhet testimi gjenetik për të vendosur një diagnozë specifike. Duke qenë një sëmundje trashëguese, anëtarët e familjes së një pacienti të konfirmuar duhet të testohen gjenetikisht, edhe nëse ata nuk kanë të njëjtat simptoma ose nuk kanë asnjë simptomë, pasi mund ta mbajnë variacionin gjenetik dhe ta transmetojnë atë te fëmijët e tyre.
PASOJAT
Përmirësimet në stilin e jetesës që nevojiten për individët me sindromën APDS 3-KD varen nga serioziteti i simptomave individuale që ka secili individ. Ekspozimi ndaj baktereve dhe viruseve të mundshme që mund të shkaktojnë infeksione mund të zvogëlohet me praktikimin e higjienës personale të mirë, kujdesin për ushqimin dhe mjedisin e jetesës; duke marrë masa paraprake gjatë notit (shmangia e liqeneve/pishinave), minimizimi i kontaktit me papastërti, tokë ose kafshë; dhe shmangia e kontaktit me njerëz që kanë infeksione. Shëndeti i pacientëve duhet të monitorohet rregullisht, duke përfshirë kontrollime për nyje limfatike të ënjtura dhe shenja e simptoma të mundshme të limfomës që janë karakteristike, siç janë: temperaturë e lartë, djersitje natën, lodhje, kruajtje dhe humbje të paqartë në peshë.
TRAJTIMI
Trajtimi për sindromën APDS 3-KD është i fokusuar në menaxhimin e simptomave, parandalimin e infeksioneve dhe zvogëlimin e inflamacioneve. Sindroma trajtohet me përdorimin e një kombinimi të terapive zëvendësuese të imunitetit me imunoglobulin për të mbështetur sistemin imunitar dhe medikamente imunosupresive për të ndihmuar në simptomat që shkaktohen nga autoimuniteti dhe inflamacioni. Për më tepër, individët me sindromën APDS 3-KD shpesh kanë nevojë për antibiotikë të përditshëm për të ndihmuar në parandalimin e infeksioneve para se ato të ndodhin. Fëmijët me sindromën APDS 3-KD mund të kenë nevojë për tuba të veshëve (tuba timpanostomi) për shkak të infeksioneve të shpeshta të veshit. Disa mund të kenë nevojë për heqjen e bajameve për shkak të ënjtjes dhe infeksioneve të sinuseve. Personat me një formë më të rëndë të sindromës APDS 3-KD mund të kenë nevojë për mbështetje respiratore, përfshirë oksigjen shtesë dhe fizioterapi për mushkëritë. Për më tepër, terapia fizike, profesionale dhe/ose e të folurit mund të ndihmojë në trajtimin e disa nga komplikimet afatgjata. Transplantimi i qelizave staminale hematopoetike (HSCT) përdoret për trajtimin e disa individëve me sindromën APDS 3-KD që nuk përgjigjen ndaj terapisë standarde. Qelizat staminale hematopoetike zhvillohen në palcën e kockave dhe kanë potencialin të bëhen çdo lloj qelize të gjakut. HSCT përfshin zëvendësimin e palcës së kockave me qeliza staminale të reja, të cilat mund të jenë qeliza imunitare funksionale dhe të shëndetshme. HSCT përdoret si trajtim për disa deficenca imunitare primare, si dhe për disa lloje të kancerit të gjakut.