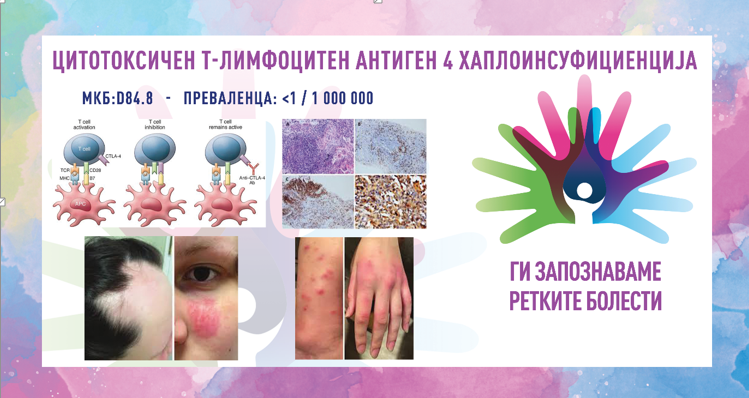
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
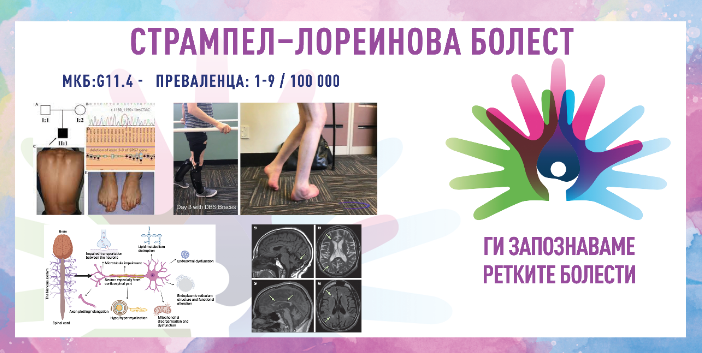
ХЕРЕДИТАРНА СПАСТИЧНА ПАРАПЛЕГИЈА/ СТРАМПЕЛ–ЛОРЕИНОВА БОЛЕСТ/ HEREDITARY SPASTIC PARAPLEGIA/ STRÜMPELL–LORRAIN DISEASE
MKБ-10 G11.4
ПРЕВАЛЕНЦА – забележана кај 1–9/100 000 население; во Европа се движи од 1/11 000 до 1/77 000 население.
ВОВЕД
Хередитарна спастична параплегија (ХСП), позната и како Стрампел–Лореинова болест, претставува високо хетерогена група на наследни болести чија главна карактеристика е прогресивно нарушување на одењето. Болеста се манифестира со прогресивна вкочанетост (спастичност) и контракција на долните екстремитети. Симптомите се резултат на дисфункција на долгите аксони во ᾿рбетниот мозок. Зафатените клетки се примарните моторни неврони, па затоа се работи за болест на горниот моторен неврон. Болеста е предизвикана од дефекти во транспортот на протеините, структурните протеини, протеините за одржување на клетките, липидите и другите супстанции низ клетката. Долгите нервни влакна (аксоните) се засегнати бидејќи долгите растојанија ги прават нервните клетки особено чувствителни на дефекти во споменатите механизми. Започнува во детството или раната адолесценција и прогресира бавно во текот на животот. Болеста првпат била опишана во 1880 година од страна на германскиот невролог Адолф Штрумпел, додека поопширно е опишана во 1888 година од страна на францускиот лекар Морис Лорен. Терминот „хередитарна спастична параплегија“ бил измислен од страна на Анита Хардинг во 1983 година.
ЕТИОЛОГИЈА
Стрампел–Лореиновата болест се должи на дисфункција на горните моторни неврони на кортикоспиналниот тракт, пренесувајќи се на наследен начин, најчесто автосомно доминантно, но може да биде и автосомно рецесивно. Тоа значи дека болеста може да се наследи од еден или двајца родители, зависно од типот на мутацијата. Најчесто е поврзана со мутации во гените SPAST (2p22.3), ATL1 (14q22.1), REEP1 (2p11.2) и KIF5A (12q.13.3) за автосомно доминантни HSPs и SPG7 (16q24.3), SPG11 (15q21). 1), и CYP7B1 (8q12.3) за автосомно рецесивен, кои кодираат протеини важни за одржување на нормалната функција на моторните неврони во спиналниот мозок. Досега се идентификувани повеќе од 80 гени одговорни за настанокот на болеста. Кодираните протеини се вклучени во многу процеси, вклучувајќи аксонален транспорт, миелинизација, трговија со ендомембрани, функции на митохондриите, комплексен липиден и нуклеотиден метаболизам. Овие мутации доведуваат до дегенерација на кортикоспиналните неврони и оштетување на спиналните нервни патишта, со што е нарушена трансмисијата потребна за контрола на движењата на мускулите на долните екстремитети. Сепак, значителен број пациенти се без генетска дијагноза по спроведено систематско тестирање.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Хередитарна спастична параплегија се манифестира со прогресивно влошување на мускулната сила во нозете, како и со зголемена спастичност. Главните симптоми вклучуваат:
спастичност – уште во раниот стадиум на болеста, пациентите почнуваат да забележуваат зголемена мускулна напнатост и тешкотии во движечките активности, како што се одењето или подигнувањето на нозете;
спастичност на долните екстремитети – постепено слабеење на мускулите на нозете, што доведува до ограничена мобилност или до потреба од помагала за одење;
ефекти во координацијата и рамнотежата – рамнотежата на телото е нарушена, а пациентите можат да имаат тешкотии во одењето, што води до чести падови;
болка во мускулите и зглобовите – може да се јави хронична болка поради мускулната напнатост;
прогресивно влошување – болеста обично има бавен прогресивен тек, но може да варира во зависност од конкретната генетска варијанта и возраст на појавата.
Клинички, херидитарните спастични параплегии можат да се поделат на чиста и комплицирана (сложена) форма.
Чистата форма на херидитарната спастична параплегија се карактеризира со бавно прогресивна спастичност и слабост на екстремитетите, најчесто поврзана со уринарни нарушувања и длабоки сензорни аномалии (намалена чувствителност на вибрација на долните екстремитети).
Сложената форма на херидитарната спастична параплегија се одликува со присуство или отсуство на дополнителни невролошки карактеристики. Невролошките карактеристики можат да вклучуваат нарушувања на церебелумот (атаксија, нистагмус, тремор), демиелинизирачка периферна невропатија (сензорни и/или моторни нарушувања), когнитивни нарушувања, сензорни дефекти, епилепсија, миопатски карактеристики, екстрапирамидални карактеристики (Паркинсонизам, хореа, дистонија), психијатриски нарушувања итн., кои можат да бидат индикативни за одреден генетски поттип. Отсуството на невролошките манифестации вклучува офталмолошки абнормалности (катаракта, ретинитис пигментоза, макуларна дегенерација) и ортопедски абнормалности (сколиоза, дислокација на зглобот и различни деформитети на стапалото).
Дијагнозата се поставува врз основа на клиничките симптоми, невролошкиот преглед, прогресивниот тек на болеста, мерењето на биомаркери, MРИ на мозокот и ᾿рбетот (со цел да се исклучат другите чести невролошки состојби), фамилијарната историја, молекуларното генетско тестирање и исклучувањето на другите диференцијални дијагнози, како што се: мултиплекс склероза, спинални васкуларни абнормалности, дефицит на витамин Б12, инфекција со HTLV-I, примарна латерална склероза, диплегична церебрална парализа, метаболички генетски заболувања, нарушување на акумулацијата на метали во мозокот. Крајната потврда на дијагнозата може да се обезбеди само со спроведување на генетски тестови насочени кон познати генетски мутации. Особено генетско тестирање е потребно во случаите каде што претходно била идентификувана мутација во семејството.
ПОСЛЕДИЦИ
Поголемиот дел од лицата со хередитарна спастична параплегија имаат нормален животен век. Симптомите на Стрампел–Лореиновата болест прогресивно се влошуваат со текот на времето, што може да доведе до ограничена мобилност, зголемен ризик од повреди, потреба од помагала. Постојат и компликации, односно хронична болка, деформации на екстремитетите и психосоцијални последици. Прогнозата зависи од фенотипот, генотипот, како и од самата експресија на гените. Во некои случаи, засегнатите лица се сериозно оневозможени, додека во други, засегнатите се способни да ги извршуваат повеќето секојдневни активности без потреба од прилагодувања.
ТРЕТМАН
Засега не постои специфичен третман кој би го спречил, забавил или би го променил текот на болеста. Терапијата е симптоматска и третманите се насочени кон ублажување на симптомите и подобрување на квалитетот на живот преку: физиотерапија и рехабилитација, медикаментозен третман (лекови против спастичност, аналгетици за ублажување на болката), хируршка интервенција (во некои случаи, за корекција на деформации), психолошка поддршка и слично. Од лекови, се препорачуваат:
баклофен – мускулен релаксант за релаксација на мускулите и намалување на тонусот, кој може да се администрира орално или интратекално;
дијазепам и клоназепам – за намалување на интензитетот на спазмите;
оксибутинин хлорид и толтеродин тартарат – неволен мускулен релаксант и спазмолитички агенс, кој се користи за намалување на спастичноста на мочниот меур кај пациенти со проблеми со контролата на мочниот меур;
кросистем – за намалување на мускулната прекумерна активност;
антидепресиви – за пациенти кои доживуваат клиничка депресија.
Особено важна е физикалната терапија која помага во обновувањето и одржувањето на способноста за движење, исто така помага за намалување на мускулниот тонус, за одржување или подобрување на опсегот на движење и мобилност, за да се зголеми силата и координацијата, за да се спречат компликации, како што се замрзнати зглобови, контрактури или рани.
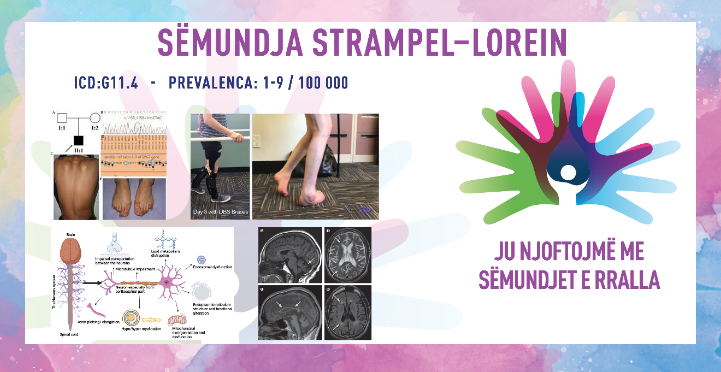
PARAPLEGJIA E TRASHËGUAR SPASTIKE/ SËMUNDJA STRAMPEL–LOREIN/ HEREDITARY SPASTIC PARAPLEGIA/ STRÜMPELL–LORRAIN DISEASE
ICD-10 G11.4
PREVALENCA – paraqitet te 1–9/100 000 banorë; në Evropë haset nga 1/11 000 deri në 1/77 000 banorë.
HYRJE
Paraplegjia e trashëguar spastike (HSP), e njohur gjithashtu si sëmundja Strampel-Lorein, është grup shumë heterogjen i sëmundjeve trashëguese, tipari kryesor i të cilave është çrregullim progresiv në ecje. Sëmundja manifestohet me ngurtësi progresive (spasticitet) dhe tkurrje të ekstremiteteve të poshtme. Simptomat janë si rezultat i mosfunksionimit të aksoneve të gjata në palcën kurrizore. Qelizat e prekura janë neuronet motorike primare, kështu që është sëmundje e neuronit të sipërm motorik. Sëmundja shkaktohet nga defekte në transportin e proteinave, proteinave strukturore, proteinave të mirëmbajtjes së qelizave, lipideve dhe substancave të tjera në të gjithë qelizën. Fijet e gjata nervore (aksonet) preken sepse distancat e gjata i bëjnë qelizat nervore veçanërisht të ndjeshme ndaj defekteve në mekanizmat e përmendur. Fillon në fëmijëri ose në adoleshencën e hershme dhe përparon ngadalë gjatë gjithë jetës. Sëmundja u përshkrua për herë të parë në vitin 1880 nga neurologu gjerman Adolf Strümpel, ndërsa u përshkrua më gjerësisht në vitin 1888 nga mjeku francez Maurice Lorrain. Termi “paraplegji e trashëguar spastike” u mendua nga Anita Harding në vitin 1983.
ETIOLOGJIA
Sëmundja Strampel-Lorein ndodh për shkak të mosfunksionimit të neuroneve të sipërme motorike të traktit kortikospinal, i transmetuar në mënyrë të trashëguar, zakonisht në mënyrë autosomale dominante, por mund të jetë edhe autosomale recesive. Kjo do të thotë se sëmundja mund të trashëgohet nga njëri ose të dy prindërit, në varësi të llojit të mutacionit. Më së shpeshti shoqërohet me mutacione në gjenet SPAST (2p22.3), ATL1 (14q22.1), REEP1 (2p11.2) dhe KIF5A (12q.13.3) për HSP-të autosomale dominante dhe SPG7 (16q24.3), SPG11. (15q21) . 1), ndërsa CYP7B1 (8q12.3) për sëmundjen autosomale recesive, të cilat kodojnë proteina të rëndësishme për ruajtjen e funksionit normal të neuroneve motorike në palcën kurrizore. Deri më tani janë identifikuar më shumë se 80 gjene përgjegjëse për shfaqjen e sëmundjes. Proteinat e koduara janë të përfshira në shumë procese, duke përfshirë transportin aksonal, mielinizimin, shkëmbimin e endomembranës, funksionet mitokondriale, metabolizmin kompleks të lipideve dhe nukleotideve. Këto mutacione çojnë në degjenerim të neuroneve kortikospinale dhe dëmtim të rrugëve nervore spinale, duke ndërprerë transmetimin e nevojshëm për të kontrolluar lëvizjet e muskujve të gjymtyrëve të poshtme. Megjithatë, një numër i konsiderueshëm i pacientëve janë pa diagnozë gjenetike pas analizave sistematike.
SIMPTOMAT DHE DIAGNOSTIKIMI
Paraplegjia e trashëguar spastike manifestohet me përkeqësim progresiv të forcës së muskujve të këmbëve, si dhe rritje të spasticitetit. Simptomat kryesore përfshijnë:
spasticitetin – tashmë në fazën e hershme të sëmundjes, pacientët fillojnë të vërejnë tension të shtuar të muskujve dhe vështirësi në aktivitetet e lëvizjes, si ecja ose ngritja e këmbëve;
spasticitetin e ekstremiteteve të poshtme – dobësim gradual i muskujve të këmbëve, që çon në lëvizje të kufizuar ose nevojë për mjete ndihmëse për ecje;
efektet në koordinim dhe ekuilibër – ekuilibri i trupit është i çrregulluar, ndërsa pacientët mund të kenë vështirësi në ecje, gjë që çon në rënie të shpeshta;
dhimbjen e muskujve dhe kyçeve – dhimbja kronike mund të paraqitet për shkak të tensionit të rritur në muskuj;
përkeqësimin progresiv – sëmundja zakonisht ka një ecuri të ngadaltë progresive, por mund të ndryshojë në varësi të variantit specifik gjenetik dhe moshës së fillimit.
Klinikisht, paraplegjia e trashëguar spastike mund të ndahet në formën e pastër dhe formën e komplikuar (komplekse).
Forma e pastër e paraplegjisë së trashëguar spastike karakterizohet nga spasticiteti dhe dobësia progresive e ngadaltë e ekstremiteteve, zakonisht të shoqëruara me çrregullime urinare dhe anomali të thella sensore (ndjeshmëria e reduktuar ndaj dridhjeve të gjymtyrëve të poshtme).
Forma komplekse e paraplegjisë së trashëguar spastike dallohet nga prania ose mungesa e veçorive shtesë neurologjike. Karakteristikat neurologjike mund të përfshijnë çrregullime cerebelare (ataksi, nistagmus, tremor), neuropati periferike demielinizuese (çrregullime sensore dhe/ose motorike), çrregullime kognitive, defekte sensore, epilepsi, tipare miopatike, tipare ekstrapiramidale (parkinsonizëm, distoni), çrregullime psikiatrike, etj, të cilat mund të jenë tregues të një nëntipi të caktuar gjenetik. Mungesa e manifestimeve neurologjike përfshin anomalitë okulistike (katarakt, retinit pigmentoz, degjenerim makular) dhe anomali ortopedike (skoliozë, dislokime të kyçeve dhe deformime të ndryshme të këmbëve).
Diagnoza bazohet në simptomat klinike, ekzaminimin neurologjik, ecurinë progresive të sëmundjes, matjet e biomarkerëve, MRI të trurit dhe shtyllës kurrizore (për të përjashtuar gjendjet e tjera të zakonshme neurologjike), historinë familjare, testimin gjenetik molekular dhe përjashtimin e diagnozave të tjera si: skleroza multipleks, anomalitë spinale vaskulare, mungesa e vitaminës B12, infeksioni me HTLV-I, skleroza anësore primare, paraliza cerebrale diplegjike, sëmundjet gjenetike metabolike, çrregullimi i akumulimit të metaleve në tru.
Konfirmimi përfundimtar i diagnozës mund të sigurohet vetëm duke kryer teste gjenetike që synojnë mutacionet e njohura gjenetike. Në veçanti, testimi gjenetik kërkohet në rastet kur një mutacion është identifikuar më parë në familje.
PASOJAT
Shumica e personave me paraplegji të trashëguar spastike kanë një jetëgjatësi normale. Simptomat e sëmundjes Strampel-Lorein përkeqësohen në mënyrë progresive me kalimin e kohës, gjë që mund të çojë në lëvizshmëri të kufizuar, rrezik të shtuar për lëndime dhe nevojë për përdorimin e mjeteve ndihmëse për të lëvizur. Ekzistojnë edhe komplikime, siç janë dhimbja kronike, deformimet e gjymtyrëve dhe pasojat psikosociale. Prognoza varet nga fenotipi, gjenotipi, si dhe nga vetë shprehja e gjenit.
Në disa raste, individët e prekur janë me aftësi të kufizuara të rënda, ndërsa në të tjera raste, individët e prekur janë në gjendje të kryejnë shumicën e aktiviteteve të përditshme pa pasur nevojë për ndihmë.
ТRAJTIMI
Aktualisht, nuk ka një trajtim specifik që do të parandalonte, ngadalësonte ose ndryshonte rrjedhën e sëmundjes. Terapia është simptomatike dhe trajtimet synojnë zbutjen e simptomave dhe përmirësimin e cilësisë së jetës nëpërmjet: fizioterapisë dhe rehabilitimit, trajtimit medikamentoz (barna kundër spasticitetit, analgjezikë për qetësimin e dhimbjeve), ndërhyrje kirurgjikale (në disa raste për korrigjimin e deformimeve), mbështetje psikologjike dhe të ngjajshme. Nga barnat rekomandohen:
baclofen – relaksues muskulor për relaksimin e muskujve dhe reduktimin e tonusit, i cili mund të administrohet në mënyrë orale ose intratekale;
diazepam dhe clonazepam – për të zvogëluar intensitetin e spazmave;
oxybutynin chloride dhe tolterodine tartrate – (agjentët antikolinergjikë) relaksues i pavullnetshëm i muskujve dhe agjent antispazmatik, që përdoren për të reduktuar spasticitetin e fshikëzës urinare te pacientët me probleme të kontrollit të fshikëzës urinare;
crosystem – për të reduktuar aktivitetin e shtuar të muskujve;
antidepresivë – për pacientët që përjetojnë depresion klinik.
Terapia fizikale është veçanërisht e rëndësishme për të ndihmuar rivendosjen dhe ruajtjen e aftësisë për të lëvizur, ajo gjithashtu ndihmon në uljen e tonusit të muskujve, për të ruajtur ose përmirësuar diapazonin e lëvizjeve dhe lëvizshmërinë, për të rritur forcën dhe koordinimin, për të parandaluar komplikimet, si nyjet e ngrira, kontraktimet ose plagët.