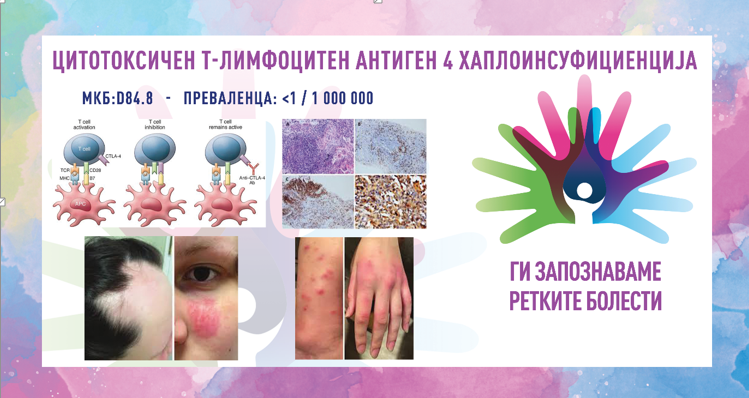
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
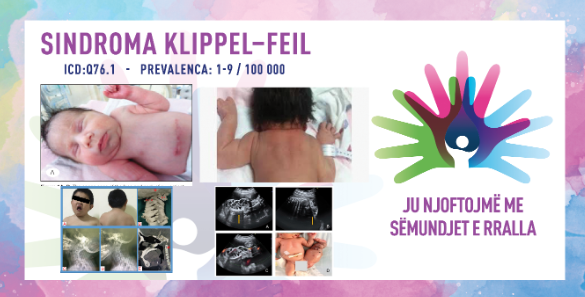
КЛИПЕЛ–ФЛЕИЛОВ СИНДРОМ/СИНДРОМ НА ЦЕРВИКАЛНА ФУЗИЈА НА ПРШЛЕНИТЕ / KLIPPEL–FEIL SYNDROME
MKБ-10 Q76.1
ПРЕВАЛЕНЦА – забележана кај 1/40.000 до 1/42.000 новороденчиња низ светот.
ВОВЕД
Клипел–Флеиловиот синдром, познат и како синдром на цервикална фузија на пршлените, е ретка конгенитална состојба која се карактеризира со абнормална фузија на која било од седумте коски на вратот (вратни пршлени). Може да резултира со ограничена способност за движење и краткост на вратот. Болеста е присутна и се дијагностицира при раѓање, со исклучок на поблаги случаи каде се дијагностицира подоцна при пројавување или влошување на симптомите. Повеќето луѓе имаат само еден или два од тие симптоми кои не се забележливи без медицинска слика. Присутно е искривување на ᾿рбетот – сколиоза, спина бифида – нецелосно формирање на ᾿рбетот за време на бременоста, односно бебето се раѓа со нецелосно споени ᾿рбетни пршлени и други скелетни абнормалности. Нарушувањето, исто така, може да биде поврзано со абнормалности на главата и лицето, скелетот, половите органи, мускулите, мозокот и ᾿рбетниот мозок, рацете, нозете и прстите. Болеста првично била пријавена во 1884 година од страна на Морис Клипел и Андре Феил од Франција. Во 1919 година, во својата докторска теза, Андре Феил предложил друга класификација на синдромот, која опфаќа не само деформација на цервикалниот ᾿рбет, туку и деформација на лумбалниот и торакалниот ᾿рбет.
ЕТИОЛОГИЈА
Кај најголем дел од лицата со Клипел–Флеиловиот синдром, болеста се појавува спорадично, односно од непознати причини. Но, во други случаи постои и наследен фактор, автосомно доминантно или автосомно рецесивно наследување. Мутациите на гените GDF6, GDF3 и MEOX1 се поврзани со синдромот. Мутациите на GDF6, GDF3 и MEOX1 предизвикуваат намален број на функционални протеини кои се кодирани од страна на овие гени, но не е јасно како недостигот на овие протеини доведува до нецелосно одвојување на пршлените кај луѓето со Клипел–Флеиловиот синдром. GDF6 и GDF3 му даваат на телото инструкции за создавање на протеини вклучени во регулирањето на растот и созревањето на коските и ᾿рскавицата. GDF6 конкретно е вклучен во формирањето на вертебралните коски, меѓу другото, и воспоставувањето на граници помеѓу коските во скелетниот развој. GDF3 е вклучен во растот на коските и ᾿рскавицата. Кога синдромот е предизвикан од мутации во гените GDF6 или GDF3, тој се наследува во автосомно доминантен модел, што значи дека една копија од изменетиот ген во секоја клетка е доволна за да предизвика нарушување. Протеинот произведен од генот MEOX1, наречен homeobox-протеин MOX-1, го регулира процесот што започнува со одвојување на пршлените еден од друг за време на раниот развој. Кога овој синдром е предизвикан од мутации во генот MEOX1, тој се наследува во автосомно рецесивен модел, што значи дека и двете копии на генот во секоја клетка имаат мутации.
Мутациите можат да се наследат на два начина:
автосомно доминантно наследување, каде што една копија од изменетиот ген во секоја клетка е доволна за да предизвика нарушување, и е поврзано со фузијата на C2-C3;
автосомно рецесивно наследување, каде што и двете копии на ген содржат мутации, и е поврзано со фузијата на C5-C6.
Идентификувана е друга автосомно доминантна форма (мапирана на локус 8q22.2), позната како Клипел–Флеилов синдром со малформација на ларинксот.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Клипел–Флеиловиот синдром се јавува во три форми, од кои е познато тека тип 1 се јавува спорадично, а тип 2 се јавува автосомно доминантно, а тип 3 се јавува автосомно рецесивно. Кај тип 1 постои фузија на најголем дел од или на цел цервикален ᾿рбет, кај тип 2, фузијата е на еден или два прешлена од цервикалниот ᾿рбет и кај тип 3 се јавуваат истите промени од тип 1 или тип 2, но придружени со фузија во торакалниот и/или лумбарниот дел од ᾿рбетот. Обично се дијагностицира по раѓањето. Специфичните симптоми варираат од пациент до пациент, но синдромот се карактеризира со тријада симптоми кои вклучуваат ограничена подвижност на вратот и горниот дел од ᾿рбетот, како и пократок врат. Најновите истражувања покажуваат дека оваа тријада ја немаат сите пациенти што значи дека кај некои се манифестира дел од овие симптоми, кај други сите симптоми. Покрај тријадата, други знаци кои се јавуваат се:
фузија на две или повеќе ᾿рбетни коски во вратот, краток врат и ниско поставена коса;
тортиколис – главата е наведната на едната страна, а брадата е свртена на другата страна;
спина бифида – вродено нарушување предизвикано од нецелосно затворање во матката на невралната туба која го обвива ᾿рбетниот мозок;
главоболки, болки во вратот и појава на испакнување на вратот поради позицијата на скапулата;
делумно неразделени прсти на раката со изглед кој асоцира на мрежа на пајак (синдактилија), нецелосен развој на скапулата (лопатките на рамото), а со тоа и ипсилатерално ограничени движења на раката;
синкинезија – движење на огледало, при кое движењето во едната рака неволно го имитира намерното движење на другата рака;
малформации на бубрезите, ребрата и срцето;
појава на бубрег во форма на потковица (horse-shoe kidney) кој се развива во ембрионалниот период, односно двата бубрега се спојуваат во еден, па се добива бубрег со форма на потковица;
респираторни проблеми;
невролошки дефицити;
проблеми со сетилото за слух и сетилото за вид.
Дијагностичката евалуација започнува при раѓањето преку длабинска клиничка евалуација, идентификација на карактеристичните симптоми и специјализирани тестови. Дијагностицирањето вклучува РТГ-тестови, компјутеризирана томографија и магнетна резонанца кои се користат за да се идентификуваат отворените простори помеѓу одредени цервикални и други пршлени или други абнормалности во коските и зглобовите. Можат да се направат и други специјализирани тестови за да се идентификуваат другите карактеристични абнормалности поврзани со состојбата, како што се оштетувања на слухот или срцеви мани. Важно е и генетското тестирање, во кое се користи примерок од плунката на детето за да се идентификува постоењето на синдромот, а EOС-технологијата за сликање која создава 3-димензионални модели од две рамни слики, овозможува подобрена дијагноза поради позиционирање. Треба да се внимава на диференцијалната дијагноза која вклучува хируршка историја на спинална фузија, анкилозен спондилитис, јувенилен ревматоиден артритис, фибродисплазија осификанс прогресива и активен или „изгорен“ остеомиелитис.
ПОСЛЕДИЦИ
Прогнозата за повеќето засегнати лица со Клипел–Флеиловиот синдром е добра доколку нарушувањето се третира рано и соодветно. Со тимски лекарски пристап според потребите и тегобите на пациентот, може да се има добра контрола врз здравствената состојба на луѓето кои го имаат овој синдром. Споените пршлени можат да го ограничат опсегот на движење на вратот и грбот, како и да доведат до хронични главоболки и болки во мускулите на вратот и грбот. Лицата со минимална зафатеност на коските често имаат помалку проблеми во споредба со лицата кои имаат само неколку погодени пршлени. Скратениот врат може да предизвика мала разлика во големината и обликот на десната и левата страна на лицето (асиметрија на лицето).
Во помалку од 30% од случаите, кај лицата со синдромот ќе се појават и срцеви мани. Доколку се присутни овие срцеви мани, тие често доведуваат до скратен животен век, во просек од 35–45 години кај мажите и 40–50 кај жените. Оваа состојба е слична на срцевата слабост забележана кај гигантизмот. Треба да се избегнуваат активности кои можат да го повредат вратот бидејќи можат да придонесат за дополнителни оштетувања. Другите болести поврзани со синдромот можат да бидат фатални ако не се лекуваат навреме или ако доцна се дијагностицираат.
ТРЕТМАН
Третманот се заснова врз управување со најчесто поврзаните симптоми, кои вклучуваат болка во вратот, радикулопатија и/или миелопатија. Основата на третманот за пациенти со болка во вратот се конзервативните, неоперативни мерки. Радикулопатијата се третира со конзервативни интервенции и во одредени случаи може да вклучува спинални инјекции. Појавата на миелопатија поради компресија на ᾿рбетниот мозок типично се припишува на вродената цервикална стеноза, но може да се влоши секундарно при компресија на ᾿рбетниот мозок со комбинација на вертебрални остеофити и/или компресија на меките ткива. Пациентите со екстензивна конгенитална фузија на грлото на матката и/или прекумерно движење на сегмент кој не е споен со движењето, се смета дека се изложени на висок ризик за повреда на ᾿рбетниот мозок. Тие треба да имаат разни секојдневни активности за да ја ограничат таквата потенцијална повреда. Хируршката интервенција може да биде оправдана за специфични случаи каде што прекумерното цервикално или краниовертебрално движење се смета за потенцијално нестабилно и е поврзано со зголемен ризик од повреда на ᾿рбетниот мозок.
SINDROMA KLIPPEL–FEIL/SINDROMA E FUSIONIT CERVIKAL / KLIPPEL–FEIL SYNDROME
ICD-10 Q76.1
PREVALENCA – vërehet në 1/40,000 deri në 1/42,000 të sapolindur në mbarë botën.
HYRJE
Sindroma Klippel–Fleil, e njohur gjithashtu si sindroma e shkrirjes vertebrale të qafës, është gjendje e rrallë kongjenitale e karakterizuar nga shkrirja jonormale e ndonjë prej shtatë kockave në qafë (rruazat e qafës). Mund të rezultojë në lëvizje të kufizuar dhe shkurtësi të qafës. Sëmundja është e pranishme dhe diagnostikohet në lindje, me përjashtim të rasteve më të lehta ku diagnostikohet më vonë me fillimin ose përkeqësimin e simptomave. Shumica e njerëzve kanë vetëm një ose dy nga këto simptoma që nuk janë të dukshme pa një ekzaminim mjekësor. Ka një lakim të shtyllës kurrizore – skoliozë, spina bifida – formim jo i plotë i shtyllës kurrizore gjatë shtatzënisë, që do të thotë se foshnja lind me rruaza kurrizore të shkrirë jo plotësisht dhe anomali të tjera skeletore. Çrregullimi mund të shoqërohet gjithashtu me anomali të kokës dhe fytyrës, skeletit, organeve gjenitale, muskujve, trurit dhe palcës kurrizore, duarve, këmbëve dhe gishtërinjve. Sëmundja u raportua fillimisht në 1884 nga Maurice Klippel dhe André Feil të Francës. Në vitin 1919, në tezën e doktoraturës, André Feil propozoi një klasifikim tjetër të sindromës, i cili përfshinte jo vetëm deformimin e shtyllës (vertebrave) të qafës, por edhe deformimin e shtyllës kurrizore lumbare dhe torakale.
ETIOLOGJIA
Në shumicën e njerëzve me sindromën Klippel-Fleil, sëmundja shfaqet në mënyrë sporadike, domethënë për arsye të panjohura. Por në raste të tjera ka edhe një faktor trashëgues, trashëgimi autosomale dominante ose autosomale recesive. Mutacionet në gjenet GDF6, GDF3 dhe MEOX1 shoqërohen me sindromën. Mutacionet në GDF6, GDF3 dhe MEOX1 shkaktojnë një numër të reduktuar të proteinave funksionale të koduara nga këto gjene, por është e paqartë se si mungesa e këtyre proteinave çon në ndarje jo të plotë vertebrale te njerëzit me sindromën Klippel-Fleil. GDF6 dhe GDF3 i japin trupit udhëzime për prodhimin e proteinave të përfshira në rregullimin e rritjes dhe maturimit të kockave dhe kërcit. GDF6 është veçanërisht i përfshirë në formimin e kockave vertebrale, ndër të tjera, ndërsa vendosjen e kufijve midis kockave në zhvillimin e skeletit. GDF3 është i përfshirë në rritjen e kockave dhe kërcit. Kur sindroma shkaktohet nga mutacione në gjenet GDF6 ose GDF3, ajo trashëgohet në një model autosomik dominant, që do të thotë se një kopje e gjenit të ndryshuar në çdo qelizë është e mjaftueshme për të shkaktuar çrregullimin. Proteina e prodhuar nga gjeni MEOX1, e quajtur proteina homeobox MOX-1, rregullon procesin që fillon me ndarjen e rruazave nga njëra-tjetra gjatë zhvillimit të hershëm. Kur kjo sindromë shkaktohet nga mutacione në gjenin MEOX1, ajo trashëgohet në një model autosomik recesiv, që do të thotë se të dy kopjet e gjenit në secilën qelizë kanë mutacione. Mutacionet mund të trashëgohen në dy mënyra:
trashëgimia autosomale dominante, ku një kopje e gjenit të ndryshuar në çdo qelizë është e mjaftueshme për të shkaktuar çrregullimin dhe shoqërohet me shkrirjen e C2-C3;
trashëgimia autosomale recesive, ku të dyja kopjet e gjenit përmbajnë mutacione dhe shoqërohet me shkrirjen e C5-C6.
Është identifikuar një formë tjetër autosomale dominante (e përcaktuar në lokusin 8q22.2), e njohur si sindroma Klippel-Fleil me keqformim laringut.
SIMPTOMAT DHE DIAGNOSTIKIMI
Sindroma Klippel-Fleil shfaqet në tre forma, nga të cilat dihet se tipi 1 shfaqet në mënyrë sporadike, tipi 2 ndodh në mënyrë autosomale dominuese dhe tipi 3 ndodh në mënyrë autosomale recesive. Në tipin 1, ka shkrirje të pjesës më të madhe ose të gjithë shtyllës së qafës, në tipin 2, shkrirja është e një ose dy vertebrave të shtyllës së qafës dhe në tipin 3, ndodhin të njëjtat ndryshime si në tipin 1 ose tipin 2 por te shoqeruara me shkrirje torakale dhe/ose lumbare. Zakonisht diagnostikohet pas lindjes. Simptomat specifike ndryshojnë nga pacienti në pacient, por sindroma karakterizohet nga një treshe simptomash që përfshijnë lëvizshmëri të kufizuar të qafës dhe shpinës së sipërme, si dhe një qafë të shkurtuar. Hulumtimet e fundit tregojnë se jo të gjithë pacientët e kanë këtë treshe, që do të thotë se disa manifestojnë disa nga këto simptoma, ndërsa të tjerët të gjitha. Përveç treshes, shenja të tjera që shfaqen janë:
bashkimi i dy ose më shumë kockave të shtyllës kurrizore në qafë, qafës së shkurtër dhe flokëve të paketuar;
torticollis – koka është e anuar në njërën anë dhe mjekra është e kthyer në anën tjetër;
spina bifida – një çrregullim kongjenital i shkaktuar nga mbyllja jo e plotë në mitër e tubit nervor që rrethon palcën kurrizore;
dhimbja e kokës, dhimbje qafe dhe shfaqja e zgjatjes së qafës për shkak të pozicionit të skapulës;
gishta pjesërisht të pandarë në dorë me një pamje që të kujton rrjetën e merimangës (sindaktili), zhvillim jo të plotë të skapulës (thika e shpatullave) dhe kështu lëvizje të kufizuara të dorës në mënyrë të njëanshme;
synkinesia – lëvizje pasqyre, në të cilën një lëvizje në njërën dorë imiton në mënyrë të pavullnetshme një lëvizje të qëllimshme në dorën tjetër;
keqformimet e veshkave, brinjëve dhe zemrës;
shfaqja e një veshke në formë patkoi (veshka patkoi) e cila zhvillohet në periudhën embrionale, pra të dy veshkat bashkohen në një, duke rezultuar në një veshkë në formë patkoi;
probleme me frymëmarrjen;
deficite neurologjike;
probleme me shqisën e dëgjimit dhe shqisën e të parit.
Vlerësimi diagnostik fillon që në lindje përmes një vlerësimi klinik të thelluar, identifikimit të simptomave karakteristike dhe testeve të specializuara. Diagnoza përfshin rrezet X, tomografinë e kompjuterizuar dhe imazhin e rezonancës magnetike, të cilat përdoren për të identifikuar hapësirat e hapura midis disa rruazave të qafës dhe vertebrave të tjera ose anomali të tjera në kocka dhe nyje. Teste të tjera të specializuara mund të bëhen për të identifikuar anomalitë e tjera karakteristike që lidhen me gjendjen, të tilla si dëmtimi i dëgjimit ose defektet e zemrës. Testimi gjenetik, i cili përdor një mostër të pështymës së fëmijës për të identifikuar praninë e sindromës, është gjithashtu i rëndësishëm dhe teknologjia e imazhit EOS, e cila krijon modele 3-dimensionale nga dy imazhe të sheshta, lejon një diagnozë të përmirësuar për shkak të pozicionimit. Vëmendje duhet t'i kushtohet diagnozës diferenciale, e cila përfshin një histori kirurgjikale të shkrirjes kurrizore, spondilitit ankilozant, artritit reumatoid juvenil, fibrodysplasia ossificans progressiva dhe osteomielitit aktiv.
PASOJAT
Prognoza për shumicën e njerëzve të prekur nga sindroma Klippel-Fleil është e mirë nëse çrregullimi trajtohet herët dhe në mënyrë të përshtatshme. Me një qasje mjekësore ekipore sipas nevojave dhe ankesave të pacientit, mund të mbahet një kontroll i mirë mbi gjendjen shëndetësore të personave që kanë këtë sindromë. Rruazat e bashkuara mund të kufizojnë gamën e lëvizjes së qafës dhe shpinës, si dhe të çojnë në dhimbje koke kronike dhe dhimbje të muskujve në qafë dhe shpinë. Njerëzit me përfshirje minimale të kockave shpesh kanë më pak probleme në krahasim me njerëzit që kanë vetëm disa rruaza të prekura. Një qafë e shkurtuar mund të shkaktojë një ndryshim të vogël në madhësinë dhe formën e anës së djathtë dhe të majtë të fytyrës (asimetria e fytyrës). Në më pak se 30% të rasteve, njerëzit me sindromën do të zhvillojnë edhe defekte në zemër. Nëse këto defekte të zemrës janë të pranishme, ato shpesh çojnë në një jetëgjatësi të shkurtuar, mesatarisht 35-45 vjet te meshkujt dhe 40-50 te femrat. Kjo gjendje është e ngjashme me dështimin e zemrës që shihet në gjigantizëm. Aktivitetet që mund të dëmtojnë qafën duhet të shmangen pasi ato mund të kontribuojnë në dëmtime të mëtejshme. Sëmundje të tjera të lidhura me sindromën mund të jenë fatale nëse nuk trajtohen në kohë ose nëse diagnostikohen vonë.
TRAJTIMI
Trajtimi bazohet në menaxhimin e simptomave më të zakonshme të lidhura, të cilat përfshijnë dhimbjen e qafës, radikulopati dhe/ose mielopati. Baza e trajtimit për pacientët me dhimbje në qafë janë masat konservative, jo kirurgjikale. Radikulopatia trajtohet me ndërhyrje konservative dhe në raste të caktuara mund të përfshijë injeksione kurrizore. Shfaqja e mielopatisë për shkak të ngjeshjes së palcës kurrizore zakonisht i atribuohet stenozës kongjenitale të qafës , por mund të përkeqësohet dytësore pas ngjeshjes së palcës kurrizore nga një kombinim i osteofiteve vertebrale dhe/ose kompresimit të indeve të buta. Pacientët me bashkim ekstensiv kongjenital të qafës dhe/ose lëvizje të tepërt të një segmenti që nuk është i bashkuar me lëvizjen konsiderohen të jenë në rrezik të lartë për dëmtim të palcës kurrizore. Ata duhet të kenë aktivitete të ndryshme ditore për të kufizuar një dëmtim të tillë të mundshëm. Ndërhyrja kirurgjikale mund të garantohet për raste specifike kur lëvizja e tepërt e qafës ose kraniovertebrale konsiderohet potencialisht e paqëndrueshme dhe shoqërohet me një rrezik në rritje të dëmtimit të palcës kurrizore.