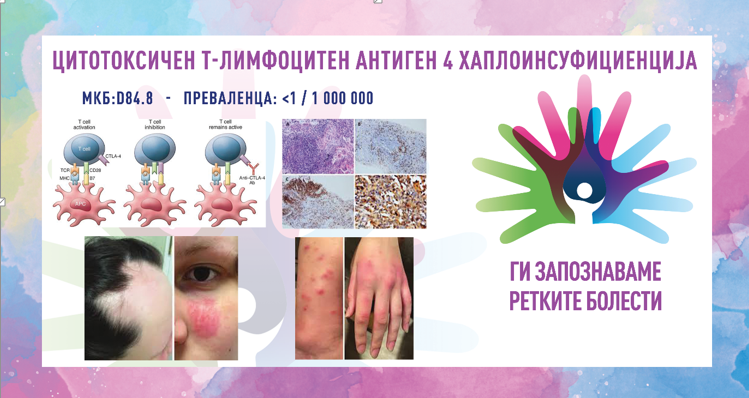
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
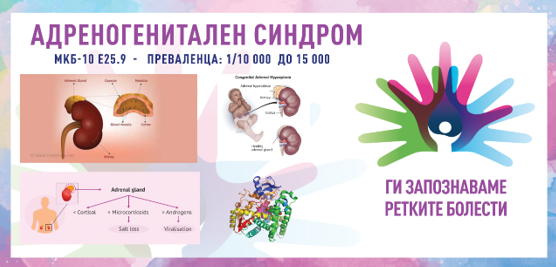
КОНГЕНИТАЛНА АДРЕНАЛНА ХИПЕРПЛАЗИЈА/ АДРЕНОГЕНИТАЛЕН СИНДРОМ/ CONGENITAL ADRENAL HYPERPLASIA (CAH)/ ADRENOGENITAL SYNDROME
MKБ-10 Е25.9
ПРЕВАЛЕНЦА – забележана кај 1/10.000 живородени деца до 1/15.000, додека кај одредени етнички групи (еврејската популација Ашкенази), таа е 1/5 000 живородени деца.
ВОВЕД
Адреногениталниот синдром, попознат како конгенитална адренална хиперплазија (CAH), е група наследни нарушувања кои ги зафаќаат надбубрежните жлезди. Овие жлезди произведуваат витални хормони, вклучувајќи кортизол, алдостерон и андрогени. CAH е резултат на ензимски дефицит кој ја нарушува синтезата на хормоните, што доведува до низа клинички манифестации во зависност од специфичниот засегнат ензим. Во 1865 година, италијанскиот лекар Луиџи де Крекио документирал случај на жена пациент со машки надворешни гениталии и зголемени надбубрежни жлезди по направена аутопсија, додека во 50-тите години забележан е значителен напредок со идентификацијата на дефицитот на 21-хидроксилаза како најчеста причина за CAH. Овој период го означил почетокот на биохемиските и хормоналните студии кои ја разјасниле патофизиологијата на болеста. Напредокот во генетската технологија кон крајот на 20-от и почетокот на 21-от век овозможува прецизна идентификација на генските мутации одговорни за CAH, подобрување на дијагнозата и овозможување скрининг на носителот.
ЕТИОЛОГИЈА
CAH е предизвикана од мутации во гените кои ги кодираат ензимите неопходни за синтезата на кортизол, алдостерон и дефицит на 21-хидроксилаза (90–95 % од случаите). Мутациите на генот CYP21A2 доведуваат до намалено производство на кортизол и алдостерон со зголемена синтеза на андрогени.
Како други ензимски недостатоци се јавуваат:
дефицит на 11β-хидроксилаза (ген CYP11B1);
недостаток на 17α-хидроксилаза (ген CYP17A1);
дефицит на 3β-хидроксистероид дехидрогеназа (ген HSD3B2);
липоиден CAH поради дефекти во стероидогениот акутен регулаторен протеин (StAR).
Недостатоците на ензими го попречуваат производството на кортизол што доведува до зголемено лачење на адренокортикотропен хормон (ACTH) поради губење на негативните повратни информации. Зголемениот ACTH го стимулира растот на надбубрежната жлезда и хиперпродукцијата на стероидни прекурсори, кои се пренасочени кон патиштата за синтеза на андрогени. Вишокот андроген резултира со вирилизација и други симптоми поврзани со андрогенот.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
1.Класичен CAH (тежок ензимски дефицит)
жени:
двосмислени гениталии при раѓање (клиторомегалија, споени усни);
нормални внатрешни репродуктивни органи.
мажи:
нормални гениталии – сепак, знаците на вишок андрогени можат рано да се појават; форма за трошење сол – симптомите се појавуваат во првите неколку недели од животот (дехидрација, повраќање, хипонатремија (низок натриум), хиперкалемија (висок калиум), хипотензија);
едноставна – вирилизирачка форма;
соодветното производство на алдостерон го спречува трошењето на сол;
вишокот на андрогени доведува до брз раст и ран пубертет.
2. Некласичен CAH (благ ензимски дефицит)
почеток – доцно детство или зрелост;
симптоми:
жени – хирзутизам, акни, нередовни менструални циклуси, неплодност;
мажи – ран развој на секундарни сексуални карактеристики, забрзан раст, но потенцијално пократка висина на возрасните поради раното затворање на епифизарите.
Дијагностицирање:
Скрининг за новороденчиња
17 нивоа на хидроксипрогестерон – покачени нивоа откриени преку тестови на крвта со убод на пети;
хормонална евалуација;
мерења на серумски хормони – зголемен 17-хидроксипрогестерон, зголемен андростендион и тестостерон;
тест за стимулација на ACTH – го проценува одговорот на надбубрежната жлезда;
анализа на електролити;
проценка за трошење сол – хипонатремија (низок натриум) и хиперкалемија (високо ниво на калиум);
генетско тестирање – идентификација на специфични генски мутации во CYP21A2 или други сродни гени;
ултразвук/МРИ – проценка на големината на надбубрежната жлезда, проценка на внатрешните репродуктивни органи во случаи на двосмислени гениталии.
ПОСЛЕДИЦИ
Напредокот во медицинската наука значително ги подобри резултатите за погодените поединци. Раната интервенција, персонализираните/лични планови за лекување и мултидисциплинарната грижа се од суштинско значење за оптимално здравје и квалитет на живот:
нормален раст и развој – повеќето поединци можат да постигнат нормална висина и развојни пресвртници;
квалитет на живот – подобрен со правилно управување со хормоналната нерамнотежа и психосоцијална поддршка;
доживотна нега – неопходен е континуиран медицински надзор за да се прилагодат третманите и да се решат компликациите.
ТРЕТМАН
Хормонска заместителна терапија
Глукокортикоиди – да се замени недостатокот на кортизол, да се потисне вишокот ACTH и да се намали производството на андрогени;
Хидрокортизон – се претпочита кај доенчиња и деца;
Преднизон или Дексаметазон – може да се користи кај адолесценти и возрасни;
Флудрокортизон – го заменува алдостеронот за да ги регулира нивоата на натриум и калиум;
додатоци на натриум – особено важни кај доенчињата.
Хируршка интервенција
хирургија за реконструкција на гениталиите – мултидисциплинарна тимска одлука која вклучува ендокринолози, хирурзи и психолози;
редовно следење – проценки за раст и развој, проценка на нивото на хормони, прилагодување на дозите на лекови по потреба;
поддршка за плодност – решавање на потенцијални репродуктивни проблеми кај возрасните;
психосоцијална поддршка – советување за пациенти и семејства;
групи за поддршка и образовни ресурси;
мерки на претпазливост за итни случаи;
дозирање на стрес – зголемени дози на глукокортикоиди за време на болест или операција за да се имитира природната реакција на телото на стрес.
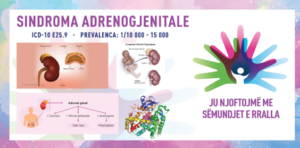
HIPERPLAZIA ADRENALE KONGJENITALE/ SINDROMA ADRENOGJENITALE/ CONGENITAL ADRENAL HYPERPLASIA (CAH)/ ADRENOGENITAL SYNDROME
ICD-10 Е25.9
PREVALENCA
Vërehet në 1/10 000 lindje të gjalla deri në 1/15 000, ndërsa në disa grupe etnike (popullata hebraike Ashkenazi), ajo është 1/5 000 lindje të gjalla.
HYRJE
Sindroma adrenogjenitale, e njohur më shumë si hiperplazia adrenale kongjenitale (CAH), është një grup çrregullimesh të trashëguara që prekin gjëndrat mbiveshkore. Këto gjëndra prodhojnë hormone jetike, duke përfshirë kortizolin, aldosteronin dhe androgjenet. CAH është rezultat i mungesës enzimatike që ndërpret sintezën e hormoneve, duke çuar në një sërë manifestimesh klinike, në varësi të enzimës së prekur specifike. Në vitin 1865 mjeku italian Luigi de Crecchio dokumentoi një rast të një pacienteje grua me gjini të jashtme mashkullore dhe gjëndra të zmadhuara mbiveshkore pas kryerjes së autopsisë, ndërsa në vitet ’50 u vërejt një përparim i rëndësishëm me identifikimin e mungesës së 21-hidroksilazës si shkaku më i zakonshëm i CAH. Kjo periudhë shënoi fillimin e studimeve biokimike dhe hormonale që shpjeguan patofiziologjinë e sëmundjes. Përparimi në teknologjinë gjenetike në fund të shekullit XX dhe fillim të shekullit XXI mundësoi identifikimin e saktë të mutacioneve gjenetike përgjegjëse për CAH, përmirësimin e diagnozës dhe mundësimin e skriningut të bartësve.
ETIOLOGJIA
CAH shkaktohet nga mutacione në gjenet që kodojnë enzimat e nevojshme për sintezën e kortizolit, aldosteronit si dhe si rezultat i mungesës së 21-hidroksilazës (90-95% e rasteve). Mutacionet në gjenin CYP21A2 çojnë në prodhim të reduktuar të kortizolit dhe aldosteronit që rezulton me rritje të sintezës së androgjeneve. Si mungesa të tjera enzimatike shfaqen:
mungesa e 11β-hidroksilazës (gjen CYP11B1);
mungesa e 17α-hidroksilazës (gjen CYP17A1);
mungesa e 3β-hidroksisteroid dehidrogjenazës (gjen HSD3B2);
CAH lipidik për shkak të defekteve në proteinën rregullatore akute steroide (StAR).
Mungesat enzimatike pengojnë prodhimin e kortizolit, gjë që çon në sekretim të shtuar të hormonit adrenokortikotropik (ACTH) për shkak të humbjes së feedback-ut negativ. ACTH i rritur stimulon rritjen e gjëndrës mbiveshkore dhe hiperprodhimin e pararendësve steroide, të cilët devijohen drejt rrugëve për sintezën e androgjeneve. Teprica e androgjeneve rezulton në virilizim dhe simptoma të tjera të lidhura me androgjenet.
SIMPTOMAT DHE DIAGNOSTIKIMI
1. CAH klasik (deficit enzimatik i rëndë)
te gratë:
gjenitale të paqarta gjatë lindjes (klitoromegali, buzë të bashkuara);
organe riprodhuese të brendshme normale.
te burrat:
gjenitale normale – megjithatë, shenjat e tepricës së androgjenit mund të shfaqen herët;
forma me humbje të kripës – simptomat shfaqen në javët e para të jetës (dehidratim, të vjella, hiponatremi (natrium i ulët), hiperkalemi (kalium i lartë), hipotension);
forma e thjeshtë – virilizim;
prodhim i duhur i aldosteronit parandalon humbjen e kripës;
teprica e androgjenit çon në rritje të shpejtë dhe pubertet të hershëm.
2. CAH joklasik (deficit enzimatik i lehtë)
fillimi – fëmijëria ose maturia e vonshme:
simptomat:
te gratë – hirsutizëm, akne, cikle menstruale të parregullta, infertilitet;
te burrat – zhvillim i hershëm i karakteristikave sekondare seksuale, rritje e përshpejtuar, por potencialisht gjatësi më e shkurtër në moshën e rritur për shkak të mbylljes së hershme të epifizave.
Diagnostikimi:
Skriningu i të sapolindurve
nivelet e 17-hidroksiprogesteronit – zbulimi i nivele të rritura përmes analizave të gjakut nga shpimi i thembrës;
vlerësim hormonal;
matjet e hormoneve serike – 17-hidroksiprogesteron i rritur, androstendion dhe testosteron të rritura;
testi i stimulimit të ACTH – vlerëson reagimin e gjëndrës mbiveshkore;
analiza e elektroliteve;
vlerësimi i humbjes së kripës – hiponatremi (natrium i ulët) dhe hiperkalemi (kalium i lartë);
testimi gjenetik – identifikimi i mutacioneve specifike në gjenin CYP21A2 ose gjene të tjera të lidhura;
ekografia/MRI – vlerësim i madhësisë së gjëndrës mbiveshkore, vlerësim i organeve të brendshme riprodhuese në raste të gjenitaleve të padefinuara.
PASOJAT
Përparimi në shkencën mjekësore ka përmirësuar ndjeshëm rezultatet për individët e prekur. Ndërhyrja e hershme, planet e personalizuara të trajtimit dhe kujdesi multidisiplinar janë thelbësore për shëndetin dhe cilësinë optimale të jetës:
rritje dhe zhvillim normal – shumica e individëve mund të arrijnë gjatësi dhe piketa zhvillimi normale;
cilësi e jetës – e përmirësuar me menaxhimin e duhur të çekuilibrimit hormonal dhe mbështetjen psikosociale;
kujdes i përjetshëm – mbikëqyrje e vazhdueshme mjekësore për të përshtatur trajtimet dhe për të adresuar ndërlikimet.
TRAJTIMI
Terapia e zëvendësimit hormonal
Glukokortikoide – për të zëvendësuar mungesën e kortizolit, për të shtypur tepricën e ACTH dhe për të ulur prodhimin e androgjenit;
Hidrokortizon – preferohet për foshnjat dhe fëmijët;
Prednizon ose Deksametazon – mund të përdoret për adoleshentët dhe të rriturit;
Fludrokortizon – zëvendëson aldosteronin për të rregulluar nivelet e natriumit dhe kaliumit;
shtesa natriumi – veçanërisht të rëndësishme për foshnjat.
Ndërhyrja kirurgjikale
kirurgjia për rindërtimin e gjenitaleve – vendim i ekipit multidisiplinar që përfshin endokrinologë, kirurgë dhe psikologë;
ndjekja e rregullt – vlerësime për rritje dhe zhvillim, vlerësim të niveleve hormonale, përshtatje të dozave të medikamenteve sipas nevojës;
mbështetja për fertilitet – adresimi i problemeve të mundshme riprodhuese te të rriturit;
mbështetje psikosociale – këshillim për pacientët dhe familjet;
grupe mbështetjeje dhe burime edukative;
masa paraprake për emergjenca;
dozimi i stresit – doza të shtuara të glukokortikoideve gjatë sëmundjeve ose operacioneve për të imituar reagimin natyral të trupit ndaj stresit.