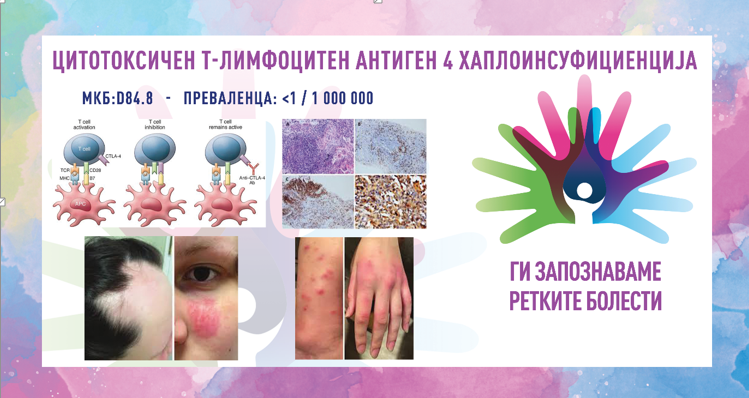
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
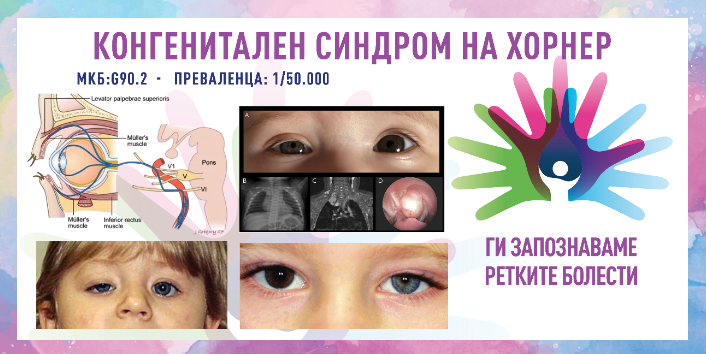
КОНГЕНИТАЛЕН СИНДРОМ НА ХОРНЕР/CONGENITAL HORNER’S SYNDROME
MKБ-10 G90.2
ПРЕВАЛЕНЦА – забележана кај 1,42/6 250 население.
ВОВЕД
Конгениталниот (вроден) синдром на Хорнер (окулосимпатична парализа) е ретко невролошко нарушување кое се карактеризира со релативна пупиларна миоза (констрикција на зеницата) и блефароптоза („овенување“ на горниот очен капак), забележливи при раѓање и предизвикани од прекин на окулосимпатичката инервација во која било точка покрај нервната патека од хипоталамусот до орбитата. Често можат да бидат присутни и дополнителни симптоми, како што се енофталм, анхидроза на лицето (отсуство на потење), хетерохромија на ирисот, конјунктивална конгестија, минлива хипотонија и/или доцнење со проширување на зеницата. Пријавена е поврзаност со траума при раѓање, неоплазми или васкуларни малформации. Кај лицата со синдромот кој се јавува пред 2-годишна возраст, обоениот дел (ирисот) на очите може да се разликува по боја (хетерохромија на ирисот) бидејќи развојот на пигментацијата (боењето) на ирисот е под контрола на цервикалните симпатички нерви и ирисот на заболеното око е посветол во боја од оној на незасегнатото око. Поединците кои развиваат синдром на Хорнер по 2-годишна возраст обично немаат хетерохромија на ирисот.
ЕТИОЛОГИЈА
Иако конгениталниот синдром на Хорнер може да се пренесе кај членови на засегнати семејства, сепак не се идентификувани поврзани гени и повеќето случаи се резултат на оштетување на нервите наречени цервикални симпатики. Овие нерви припаѓаат на делот од нервниот систем кој ги контролира неволните функции (автономниот нервен систем). Во рамките на автономниот нервен систем, нервите се дел од поделбата наречена симпатичен нервен систем. Цервикалните симпатички нерви контролираат неколку функции во окото и лицето, како што се проширување на зеницата и потење. Сепак, како најчеста причина за конгениталниот синдром на Хорнер се смета дека е траума при раѓање, операција или случајна повреда што резултира со повреда на брахијалниот плексус. Пријавени се и потенцијални причини за конгениталниот синдром на Хорнер, вклучувајќи торакален и цервикален невробластом, агенеза на внатрешната каротидна артерија и компликации од перинатални хируршки процедури, односно причини опасни по живот.
Синдромот на Хорнер може да биде предизвикан и од проблеми со артеријата која ги снабдува главата и вратот со крв (каротидната артерија), што резултира со губење на протокот на крв во нервите. Некои лица со конгениталниот синдром на Хорнер имаат недостаток на развој (агенеза) на каротидната артерија. Кинењето на слоевите на ѕидот на каротидната артерија (дисекција на каротидна артерија) исто така може да доведе до појава на синдромот.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Првите симптоми вообичаено ги забележуваат родителите кај своите деца, и тоа: разлика во бојата на очите, големината на зениците или нарушено црвенило на лицето.
Пациентите можат да се жалат и на црвенило на очите, назална затнатост, главоболка, како и многу други невролошки или системски дефицити, па затоа задолжителен е насочен физички и невролошки преглед сè со цел да се идентификуваат знаците кои можат да помогнат во локализирање на лезијата:
окуларни знаци
очни капаци – пациентите имаат блага (помалку од 2 mm) птоза на горниот капак и инверзна птоза на долниот капак (долниот капак лежи на повисоко ниво од нормалното), што предизвикува намалена палпебрална бленда;
анизокорија – пациентите кои имаат анизокорија (разлика во големината на зеницата), имаат и птотичко око, односно помала зеница (миоза); анизокоријата е поизразена во темнина, што укажува на патологијата на дилататорот на зеницата; на помалата зеница ѝ треба подолго време да се прошири кога светлиот извор на светлина ќе се оддалечи од окото; овој феномен се нарекува доцнење на дилатација;
хетерохромија на ирисот – ириси со различни бои;
екстраокуларни движења – можат да бидат присутни кај лезиите на мозочното стебло или кавернозниот синус;
други знаци на симпатична денервација вклучуваат ипсилатерална конјунктивална инјекција, промени во сместувањето и понизок интраокуларен притисок;
невролошки знаци
знаци на мозочно стебло – атаксија, диплопија, нистагмус, странична слабост или вкочанетост, засипнатост и дисфагија;
знаци на ᾿рбетен мозок (миелопатски) – сензорни или моторни абнормалности со ниво на ᾿рбетниот мозок (дисфункција на движења на дебелото црево или мочниот меур, еректилна дисфункција кај мажите и спастицитет);
брахијална плексопатија – болка и слабост во раката или рацете;
кранијалната невропатија (единечни или повеќекратни кранијални нерви) – може да биде предизвикана од лезии во кавернозниот синус или основата на черепот;
други знаци
анхидроза – може да се забележи променлив степен на губење на потењето во зависност од местото на лезијата;
Арлекинов знак – нарушено црвенило на едната половина на лицето (или полутело) забележано кај деца со симпатична денервација на лицето;
цервикална или абдоминална маса – може да се забележи кај деца со невробластом.
Некои луѓе кои го имаат синдромот немаат ниту еден познат симптом што би довел до оштетување на нервите, ниту пак историја на нарушување во нивното семејство. Овие случаи се нарекуваат идиопатски синдром на Хорнер.
Првиот чекор во дијагностицирањето на конгениталниот синдром на Хорнер е да се извршат соодветни клинички анализи за да се идентификуваат причините кои довеле до појава на синдромот. Почетните анализи вклучуваат набљудување на птоза (на горните и долните капаци), миоза на птотичкото око и демонстрација на заостанување на дилатацијата во заболеното око и анхидроза на истата страна. Невролошките абнормалности можат да се потврдат со студии за нервната спроводливост и електромиографија на зафатениот екстремитет. Потребна е радиолошка евалуација на мозокот, цервикалниот ᾿рбетен мозок, церебралната васкулатура, главата, вратот и градниот кош. Најчесто се применува конвенционалниот ангиограм, додека фармаколошките тестови за потврда можат да се направат во суптилни случаи или кога се сомневаме на некоја диференцијална дијагноза.
ПОСЛЕДИЦИ
Абнормалностите во пределот на очите поврзани со конгениталниот синдром на Хорнер генерално не влијаат на видот или здравјето. Сепак, оштетувањето на нервите кое го предизвикува синдромот на Хорнер, може да резултира со други здравствени проблеми, а некои можат да бидат опасни по живот, па затоа соодветната евалуација и навремената дијагноза можат да го спасат животот на засегнатото лице.
ТРЕТМАН
Третманот кај пациентите е симптоматски и зависи од етиологијата која довела до настанување на синдромот. Третманот вклучува:
дисекција на каротидна артерија – бидејќи пациенти се изложени на зголемен ризик за церебрален инфаркт, тие треба веднаш да се лекуваат со антикоагулација под надзор на невролог;
третман на невробластом – децата кои го имаат синдромот без очигледна причина, како што е траума, треба да се проценат за системски малигнитет, особено невробластом, со помош на педијатар; тие треба да се евалуираат за маси на вратот и абдоменот и да се тестираат за уринарни катехоламински метаболити; потребна е соодветна радиолошка евалуација на главата, вратот и градниот кош која треба да се добие во консултација со педијатар;
третман на блефароптоза – хируршкиот пристап варира во зависност од преференциите на хирургот; бидејќи функцијата на леваторната палпебра е нормална, операцијата обично вклучува зајакнување на дејството на леваторниот мускул; пристапот може да вклучува или апоневротично напредување или мускулна конјуктивална ресекција на Милер.
Дополнително, ако примарните индикации за третман на која било форма на вродена птоза се амблиопија и абнормално позиционирање на главата, тогаш се советува рана хируршка интервенција. Тешката билатерална (или еднострана) птоза може да предизвика пациентот да заземе очигледна положба на главата под брадата што може да го попречи нормалното функционирање. Исто така, корекцијата на птозата може да се одложи додека пациентот не стане доволно возрасен за операцијата да се изврши само со локална анестезија бидејќи тоа овозможува интраоперативни прилагодувања. Ако на пациент со птоза му е потребна и операција на страбизам, операцијата на вертикалните мускули може да ја промени положбата на очните капаци. Затоа, операцијата за птоза често се одложува додека не се коригира страбизмот.
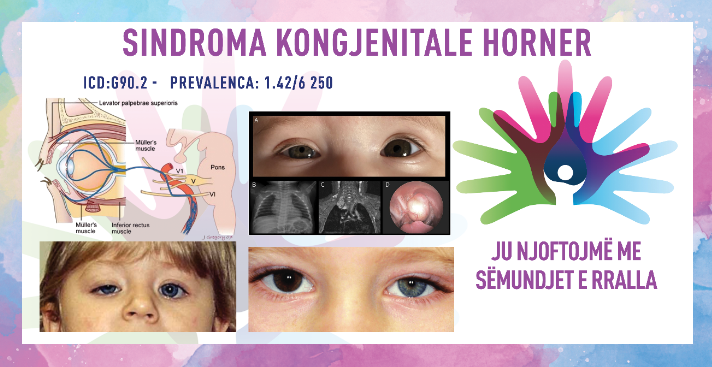
SINDROMA KONGJENITALE HORNER/ CONGENITAL HORNER’S SYNDROME
ICD-10 G90.2
PREVALENCA – vërejtur në 1.42/6 250 popullatë.
HYRJE
Sindroma kongjenitale Horner (paraliza okulosimpatike) është çrregullim i rrallë neurologjik i karakterizuar nga mioza relative e pupilës (shtrëngimi i bebëzës) dhe blefaroptoza (rënia e qepallës së sipërme), e dukshme në lindje dhe e shkaktuar nga ndërprerja e inervimit okulosimpatik në çdo pikë përgjatë nervit. rruga nga hipotalamusi në orbitë. Shpesh mund të jenë të pranishme simptoma të tjera, të tilla si enophthalmos, anhidroza e fytyrës (mungesa e djersitjes), heterokromia e irisit, kongjestion i konjuktivës, hipotonia kalimtare dhe/ose zgjerimi i vonuar i pupilës. Është raportuar një lidhje me traumat e lindjes, neoplazitë ose keqformimet vaskulare. Tek individët me sindromën që shfaqet para moshës 2 vjeçare, pjesa e ngjyrosur (irisi) e syve mund të ndryshojë në ngjyrë (heterokromia e irisit), sepse zhvillimi i pigmentimit të irisit (ngjyrosja) është nën kontrollin e nervave simpatikë të qafës (cervikale) dhe irisi i syrit të prekur është me ngjyrë më të çelur se syri i paprekur. Individët që zhvillojnë sindromën Horner pas moshës 2 vjeçare zakonisht nuk kanë heterokromi të irisit.
ETIOLOGJIA
Edhe pse sindroma kongjenitale Horner mund të transmetohet përmes anëtarëve të familjes së prekur, nuk janë identifikuar gjene shoqëruese dhe shumica e rasteve janë rezultat i dëmtimit të nervave të quajtur simpatikë cervikale. Këto nerva i përkasin pjesës së sistemit nervor që kontrollon funksionet e pavullnetshme (sistemi nervor autonom). Brenda sistemit nervor autonom, nervat janë pjesë e një ndarjeje të quajtur sistemi nervor simpatik. Nervat simpatikë të qafës (cervikale) kontrollojnë disa funksione në sy dhe fytyrë, si zgjerimi i bebëzës dhe djersitja. Megjithatë, shkaku më i zakonshëm i sindromës kongjenitale Horner mendohet të jetë trauma e lindjes, operacioni ose lëndimi aksidental që rezulton në dëmtim të pleksusit brahial. Janë raportuar gjithashtu shkaqe të mundshme të sindromës Horner kongjenitale, duke përfshirë neuroblastomën torakale dhe të qafës (cervikale), agjenezën e arteries karotide të brendshme dhe komplikime nga procedurat kirurgjikale perinatale, d.m.th., shkaqe kërcënuese për jetën.
Sindroma Horner mund të shkaktohet edhe nga problemet me arterien që furnizon me gjak kokën dhe qafën (arteria karotide), duke rezultuar në humbjen e rrjedhjes së gjakut në nerva. Disa njerëz me sindromën kongjenitale Horner kanë mungesë të zhvillimit (agjenezës) të arteries karotide. Prishja e shtresave të murit i arteries karotide (diseksioni i arteries karotide) gjithashtu mund të çojë në sindromën.
SIMPTOMAT DHE DIAGNOSTIKIMI
Simptomat e para zakonisht vihen re nga prindërit te fëmijët e tyre, përkatësisht: një ndryshim në ngjyrën e syve, madhësinë e bebëzës ose skuqje jonormale të fytyrës.
Pacientët gjithashtu mund të ankohen për skuqje të syve, kongjestion nazal, dhimbje koke, si dhe shumë deficite të tjera neurologjike ose sistemike, kështu që një ekzaminim fizik dhe neurologjik i fokusuar është i detyrueshëm për të identifikuar shenjat që mund të ndihmojnë në lokalizimin e lezionit:
- shenja okulare
- qepallat – pacientët kanë ptozë të lehtë (më pak se 2 mm) të qepallës së sipërme dhe ptozë të anasjelltë të qepallës së poshtme (qepalla e poshtme shtrihet në një nivel më të lartë se normalja), gjë që shkakton një hapje palpebrale të reduktuar;
- anizokoria – pacientët që kanë anizokorinë (ndryshim në madhësinë e bebëzës) kanë gjithashtu një sy ptotik, pra një bebëzë më të vogël (miozë); anizokoria është më e theksuar në errësirë, që tregon për patologjinë e zgjeruesit të bebëzës; bebëzës më të vogël i duhet më shumë kohë për t’u zgjeruar kur një burim i ndritshëm drite largohet nga syri; ky fenomen quhet zgjerim i vonuar;
- heterokromia e irisit – irise me ngjyra të ndryshme;
- lëvizjet ekstraokulare – mund të jenë të pranishme në lezione të trungut të trurit ose sinusit kavernoz;
- shenja të tjera të denervimit simpatik përfshijnë injeksion konjuktival ipsilateral, ndryshime në akomodim dhe presion më të ulët intraokular;
- shenjat neurologjike
- shenjat e trungut të trurit – ataksi, diplopi, nistagmus, dobësi ose mpirje anësore, ngjirurit e zërit dhe disfagi;
- shenjat e palcës kurrizore (mielopatike) – anomalitë shqisore ose motorike në nivelin e palcës kurrizore (mosfunksionimi i lëvizjeve të zorrëve ose fshikëzës, mosfunksionimi erektil te meshkujt dhe spasticiteti);
- plexopatia brachiale – dhimbje dhe dobësi në krah ose duar;
- neuropatia kraniale (nervat kraniale të vetme ose të shumëfishta) – mund të shkaktohet nga lezione në sinusin kavernoz ose në bazën e kafkës;
- shenja të tjera
- anhidroza – mund të vërehet një shkallë e ndryshueshme e humbjes së djersitjes në varësi të vendndodhjes së lezionit;
- shenja Harlequin – skuqje jonormale e gjysmës së fytyrës (ose gjysmës së trupit) e vërejtur te fëmijët me denervim simpatik të fytyrës;
- masa cervikale ose abdominale – mund të vërehet te fëmijët me neuroblastomë.
Disa njerëz që kanë sindromën nuk kanë simptoma të njohura që do të çonin në dëmtim nervor, as histori të çrregullimit në familjen e tyre. Këto raste quhen sindroma idiopatike Horner.
Hapi i parë në diagnostikimin e sindromës kongjenitale Horner është kryerja e testeve klinike të përshtatshme për të identifikuar shkaqet që çuan në shfaqjen e sindromës. Analizat fillestare përfshijnë vëzhgimin e ptozës (të qepallave të sipërme dhe të poshtme), miozën e syrit ptotik dhe demonstrimin e zgjerimit të vonuar në syrin e prekur dhe anhidrozën në të njëjtën anë. Anomalitë neurologjike mund të konfirmohen nga studimet e përcjelljes nervore dhe elektromiografia e gjymtyrës së prekur. Kërkohet vlerësimi radiologjik i trurit, palcës kurrizore të qafës së mitrës, vaskulaturës cerebrale, kokës, qafës dhe gjoksit. Më së shpeshti përdoret angiogrami konvencional, ndërsa testet konfirmuese farmakologjike mund të kryhen në raste delikate ose kur dyshohet për një diagnozë diferenciale.
PASOJAT
Anomalitë në zonën e syve të shoqëruara me sindromën kongjenitale Horner në përgjithësi nuk ndikojnë në shikimin ose shëndetin. Megjithatë, dëmtimi nervor që shkakton sindroma Horner mund të rezultojë në probleme të tjera shëndetësore, disa prej të cilave mund të jenë kërcënuese për jetën, kështu që vlerësimi i duhur dhe diagnoza në kohë mund të shpëtojnë jetën e personit të prekur.
TRAJTIMI
Trajtimi për pacientët është simptomatik dhe varet nga etiologjia që çon në sindromën. Trajtimi përfshin:
- diseksioni të arteries karotide – për shkak se pacientët janë në rrezik të shtuar për infarkt cerebral, ata duhet të trajtohen menjëherë me antikoagulim nën mbikëqyrjen e një neurologu;
- trajtim të neuroblastomës – fëmijët që kanë sindromën pa një shkak të dukshëm, si trauma, duhet të vlerësohen për malinjitet sistemik, veçanërisht neuroblastoma, me ndihmën e një pediatri; ato duhet të vlerësohen për masat e qafës dhe barkut dhe të testohen për metabolitët e katekolaminës urinare; kërkohet një vlerësim i duhur radiologjik i kokës, qafës dhe gjoksit dhe duhet të merret në konsultim me një pediatër;
- trajtim të blefaroptozës – qasja kirurgjikale ndryshon në varësi të preferencave të kirurgut; për shkak se funksioni i palpebrave levator është normal, operacioni zakonisht përfshin forcimin e veprimit të muskulit levator; qasja mund të përfshijë ose avancimin aponeurotik ose rezeksionin muskulor konjuktival të Millerit.
Përveç kësaj, nëse indikacionet parësore për trajtimin e çdo forme të ptozës kongjenitale janë ambliopia dhe pozicionimi jonormal i kokës, atëherë këshillohet ndërhyrja e hershme kirurgjikale. Ptoza e rëndë dypalëshe (ose e njëanshme) mund të shkaktojë që pacienti të marrë një pozicion të dukshëm me kokë poshtë që mund të ndërhyjë në funksionimin normal. Gjithashtu, korrigjimi i ptozës mund të vonohet derisa pacienti të rritet mjaftueshëm që operacioni të kryhet vetëm me anestezi lokale, sepse kjo lejon rregullimet intraoperative. Nëse një pacient me ptozë gjithashtu ka nevojë për kirurgji të strabizmit, operacioni në muskujt vertikal mund të ndryshojë pozicionin e qepallave. Prandaj, operacioni i ptozës shpesh vonohet derisa strabizmi të korrigjohet.