Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
СИНДРОМ НА ЖЕНУ/ЖЕНУ-АСФИКСИЈАЛНА ТОРАКАЛНА ДИСТРОФИЈА / JEUNE SYNDROME/ JEUNE ASPHYXIATING THORACIC DYSTROFY
MKБ-10 Q77.2
ПРЕВАЛЕНЦА – забележан кај приближно 1/100.000 до 1/130.000 живородени деца.
ИНЦИДЕНЦА – од 1–5/500.000 раѓања.
ВОВЕД
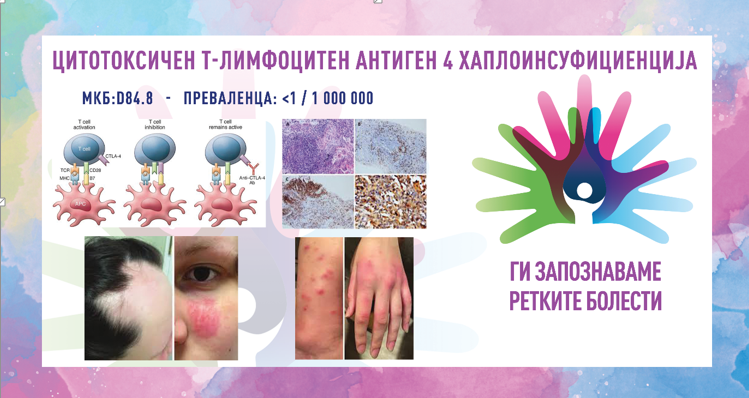
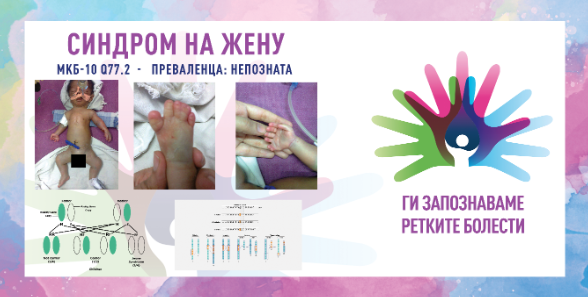
Синдромот на Жену, познат и како асфиксијален торакален дистофичен синдром, е ретка генетска болест која влијае на развојот на ребрата и градниот кош. Синдромот води до абнормален развој на градниот кош, што може да го ограничи растот на белите дробови и да предизвика респираторни проблеми. Кај новороденчињата, овој синдром често води до тешкотии со дишењето поради стеснетиот граден кош, што ја намалува способноста на белите дробови да се прошират правилно. Кај лицата со овој синдром често се појавуваат и други скелетни абнормалности, вклучувајќи скратени коски на рацете и нозете, како и проблеми со функцијата на бубрезите. Состојбата се наследува на автосомно рецесивен начин, што значи дека двајцата родители мораат да носат копија од мутираниот ген за нивното дете да биде засегнато од синдромот.
ЕТИОЛОГИЈА
Синдромот на Жену е наследно заболување кое предизвикува физички аномалии и респираторни проблеми, а главните причини за неговото развивање вклучуваат генетски мутации и наследни механизми. Молекуларната основа на синдромот е делумно разјаснета што укажува на инволвираност/вклученост на гените IFT80 (3q25.33), DYNC2H1 (11q22.3), WDR19 (4p14) и TTC21B (2q24.3), од кои секој кодира по некој интрафлагеларен транспортен протеин, кој потврдува дека овој синдром припаѓа на групата цилопатии. Мутациите во другите гени, исто така, можат да бидат вклучени во болеста и останува истите да се идентификуваат. Синдромот на Жену се наследува на автозомно-рецесивен начин, што значи дека детето мора да наследи две копии од мутираниот ген, една од секој родител, за да развие синдром. Ако само еден родител е носител на мутираниот ген, детето обично нема симптоми, но може да биде носител на мутацијата. Иако генетските мутации се главни причинители за развој на овој синдром, истражувањата покажуваат дека некои фактори од животната средина, како токсини или инфекции, играат улога во развојот на синдромот, но не е целосно докажано.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Синдромот на Жену е препознатлив во антенаталниот период или веднаш при раѓање. Во ретки случаи, може да биде присутна и постаксијална полидактилија. Тесниот граден кош може да предизвика неонатална респираторна инсуфициенција и може да биде поврзан со постојани респираторни манифестации. Некои случаи се тешки, додека други имаат бениген тек. Стапката на раст е променлива, но може да биде речиси нормална. Забележани се хепатална и бубрежна инсуфициенција во ретки случаи (фиброза на црниот дроб или нефронофтиза) кои се јавуваат на која било возраст. Може да се забележи и ретинална пигментна дегенерација. Додека пак, интелектуалниот развој е нормален.
Симптомите варираат во зависност од интензитетот и типот на синдромот:
низок раст, пократки прсти на екстремитетите или дополнителни прсти на рацете и нозете (полидактилија);
аномалии на граден кош, како што се респираторни проблеми и аномалии во структурата на ребрата кои можат да влијаат на респираторната функција;
респираторни проблеми, проблеми со дишењето поради недостаток на развој на белите дробови или хипоплазија, со што се јавува потреба за респираторна поддршка;
срцеви аномалии – многу деца со овој синдром развиваат срцеви аномалии, што може да вклучува структурни проблеми со срцето;
аномалии во развојот на бубрезите;
мускулна слабост (хипотонија) што може да влијае на моторичката способност на детето.
Дијагнозата се заснова на радиолошки наоди: кратки ребра, абнормална морфологија на карлица, со хоризонтален покрив на ацетабуларниот покрив и троен формиран аспект. Рацете се нормални или кратки со можни епифизи во форма на конус во фалангите. Диференцијалната дијагноза може да вклучува тораколарингопелвична дисплазија, Елис-ван Кревелдов синдром, Сенсенбренеров синдром и татковска еднородителска дисомија на хромозомот 14, па затоа медицинските тимови треба да внимаваат при дијагностицирањето и при донесување на конечната одлука.
ПОСЛЕДИЦИ
Прогнозата е многу променлива во зависност од висцералните асоцирани болести, а ризикот од тешки респираторни компликации се намалува по 2-годишна возраст. Децата можат да развијат висок крвен притисок, заболување на црниот дроб, цисти на панкреасот, абнормалности на забите и ретинална дистрофија, болест на очите што може да доведе до губење на видот, мускулна слабост, како и физички аномалии.
ТРЕТМАН
Третманот на овој синдром е комплексен, индивидуален и обично вклучува мултидисциплинарен пристап, со цел да се управува со симптомите и да се подобри квалитетот на животот на пациентите. Поради тоа што синдромот вклучува различни здравствени проблеми, третманот треба да биде прилагоден на специфичните потреби на секое дете. Респираторната поддршка е една од најважните аспекти на третманот. Децата со овој синдром често имаат недоволно развиени бели дробови и аномалии во градниот кош, што доведува до проблеми со дишењето. Во многу случаи, потребна е респираторна поддршка, како што се кислородна терапија или механичката вентилација. Следењето на бубрежната функција е од големо значење, а во случаи на бубрежна инсуфициенција може да се јави потреба од третман, вклучувајќи дијализа или трансплантација на бубрег. Исто така, од клучно значење се систематското следење на респираторната функција и интервенциите во зависност од потребите на детето. Многу деца со овој синдром имаат аномалии на срцето, па од голема важност е да се следи кардиоваскуларното здравје, а во некои случаи може да биде потребна хируршка интервенција за корекција на срцевите аномалии. Поради мускулната слабост и хипотонија, децата со синдромот на Жену можат да имаат потешкотии со развојот на моторичките способности, а физикалната терапија може да биде од голема помош во подобрувањето на моторичките вештини, сила и координација. Оваа терапија е од голема помош за детето да може понатаму да ги извршува секојдневните активности. Во случаи на сериозни физички аномалии, како што се деформации на екстремитетите или градниот кош, можат да се направат и оперативни интервенции. Поддршката од психолозите може да помогне во справувањето со предизвиците и во подобрувањето на самопочитта и социјалните вештини на детето. Децата можат да имаат потреба од специјализирана образовна програма или адаптација на училиште.
SINDROMA JEUNE/DISTROFIA TORAKALE ASFIZIKE JEUNE / JEUNE SYNDROME/JEUNE ASPHYXIATING THORACIC DYSTROFY
ICD-10 Q77.2
PREVALENCA – Sindroma jeune vërehet në lindje afërsisht 1/100 000 – 1/130 000
INCIDENCA – 1-5/ 500 000 lindje
HYRJE
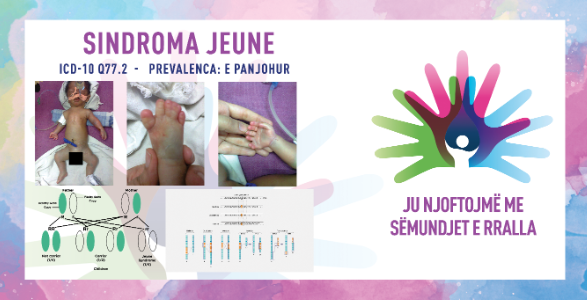
Sindroma jeune, e njohur edhe si sindroma distrofike torakale asfizike jeune, është sëmundje e rrallë gjenetike që ndikon në zhvillimin e brinjëve dhe gjoksit. Sindroma çon në zhvillim jonormal të gjoksit, i cili mund të kufizojë rritjen e mushkërive dhe të shkaktojë probleme të frymëmarrjes. Te të sapolindurit, kjo sindromë shpesh çon në vështirësi në frymëmarrje për shkak të ngushtimit të gjoksit, gjë që redukton aftësinë e mushkërive për t'u zgjeruar siç duhet. Njerëzit me këtë sindromë shpesh kanë anomali të tjera skeletore, duke përfshirë kockat e shkurtuara në krahë dhe këmbë, si dhe probleme me funksionin e veshkave. Gjendja trashëgohet në një mënyrë autosomale recesive, që do të thotë se të dy prindërit duhet të mbajnë një kopje të gjenit të mutuar që fëmija i tyre të preket nga sindroma.
ETIOLOGJIA
Sindroma jeune është sëmundje e trashëguar që shkakton anomali fizike dhe probleme të frymëmarrjes. Arsyet kryesore për zhvillimin e tij përfshijnë mutacionet gjenetike dhe mekanizmat trashëgues. Baza molekulare e sindromës është sqaruar pjesërisht duke sugjeruar përfshirjen e gjeneve IFT80 (3q25.33), DYNC2H1 (11q22.3), WDR19 (4p14) dhe TTC21B (2q24.3), secila prej të cilave kodon një proteinë transporti intraflagelar, duke konfirmuar se sindroma Jeanne i përket grupit të cilopative. Mutacionet në gjene të tjera gjithashtu mund të jenë të implikuara në sëmundje dhe mbeten për t'u identifikuar. Sindroma Geneu trashëgohet në një mënyrë autosomale recesive, që do të thotë se një fëmijë duhet të trashëgojë dy kopje të gjenit të mutuar, një nga secili prind, për të zhvilluar sindromën. Nëse vetëm njëri prind mbart gjenin e mutuar, fëmija zakonisht nuk ka simptoma, por mund të jetë bartës i mutacionit. Edhe pse mutacionet gjenetike janë shkaktarët kryesorë të zhvillimit të kësaj sindrome, disa kërkime tregojnë se faktorët mjedisorë, si toksinat apo infeksionet, luajnë një rol në zhvillimin e sindromës, por kjo nuk është plotësisht e vërtetuar.
SIMPTOMAT DHE DIAGNOSTIKIMI
Sindroma Jenu është e njohur në periudhën antenatale ose menjëherë në lindje. Në raste të rralla mund të jetë e pranishme edhe polidaktilia postaksiale. Një gjoks i ngushtë mund të shkaktojë dështim të frymëmarrjes neonatale dhe mund të shoqërohet me manifestime të vazhdueshme të frymëmarrjes. Disa raste janë të rënda, ndërsa të tjerat kanë ecuri beninje. Shkalla e rritjes është e ndryshueshme, por mund të jetë pothuajse normale. Insuficienca hepatike dhe renale është vërejtur në raste të rralla (fibrozë e mëlçisë ose nefronoftizë) që ndodhin në çdo moshë. Mund të vërehet gjithashtu degjenerim pigmentar i retinës. Zhvillimi intelektual është normal. Simptomat ndryshojnë në varësi të intensitetit dhe llojit të sindromës:
shtat i shkurtër, gishta më të shkurtër në gjymtyrë ose gishtërinj dhe gishta shtesë (polidaktili);
anomali të kraharorit, si probleme me frymëmarrje dhe anomali të strukturës së brinjëve që mund të ndikojnë në funksionin e frymëmarrjes, probleme me frymëmarrjen për shkak të mungesës së zhvillimit të mushkërive ose hipoplazisë, që rezulton në nevojën për mbështetje të frymëmarrjes;
anomali të zemrës – shumë fëmijë me sindromën Genou; zhvillimi i anomalive të zemrës, të cilat mund të përfshijnë probleme strukturore me zemrën;
anomali të në zhvillimin e veshkave;
dobësi të muskujve (hipotonia) që mund të ndikojë aftësia motorike e fëmijës.
Diagnoza bazohet në gjetjet radiologjike: brinjët janë të shkurtra dhe legeni ka një morfologji jonormale, me një çati horizontale të çatisë acetabulare dhe një aspekt trekëndor të formuar nga një fryrje mesatare dhe dy spursa anësore. Krahët janë të drejtë ose të shkurtër me epifiza të mundshme në formë koni në falangat. Diagnoza diferenciale mund të përfshijë displazi torakolaringopelvike, sindromën Ellis-van Creveld, sindromën Sensenbrenner dhe dizomi uniprindërore nga babai në kromozomin 14, prandaj duhet treguar kujdes nga ekipet mjekësore në vendosjen e diagnozës dhe marrjen e vendimit përfundimtar.
PASOJAT
Prognoza është shumë e ndryshueshme në varësi të sëmundjeve të lidhura me organet e brendshme dhe rreziku i komplikimeve të rënda respiratore zvogëlohet pas moshës 2 vjeçare. Fëmijët mund të zhvillojnë presion të lartë të gjakut, sëmundje të mëlçisë, ciste pankreatike, anomali të dhëmbëve dhe distrofi të retinës, sëmundje të syrit që mund të çojnë në humbje të shikimit, dobësi të muskujve, si dhe në anomali fizike.
TRAJTIMI
Trajtimi i sindromës jeune është kompleks, individual dhe zakonisht përfshin një qasje multidisiplinare, e gjitha me synimin për të menaxhuar simptomat dhe për të përmirësuar cilësinë e jetës së pacientëve. Për shkak se sindroma përfshin një sërë problemesh shëndetësore, trajtimi duhet t'u përshtatet nevojave specifike të çdo fëmije. Mbështetja e frymëmarrjes është një nga aspektet më të rëndësishme të trajtimit. Fëmijët me këtë sindromë shpesh kanë mushkëri të pazhvilluara dhe anomali në gjoks, të cilat çojnë në probleme me frymëmarrjen. Në shumë raste, kërkohet mbështetje respiratore, si terapia me oksigjen ose ventilim mekanik. Monitorimi i funksionit renal është i rëndësishëm dhe në rastet e dështimit të veshkave, mund të kërkohet trajtim, duke përfshirë dializën ose transplantin e veshkave. Monitorimi sistematik i funksionit të frymëmarrjes dhe ndërhyrjet në varësi të nevojave të fëmijës janë vendimtare. Shumë fëmijë me këtë sindromë kanë anomali të zemrës, ndaj është shumë e rëndësishme të monitorohet shëndeti kardiovaskular dhe në disa raste mund të kërkohet ndërhyrje kirurgjikale për të korrigjuar anomalitë e zemrës. Për shkak të dobësisë së muskujve dhe hipotonisë, fëmijët me sindromën jeune mund të kenë vështirësi në zhvillimin e aftësive motorike dhe terapia fizike mund të jetë një ndihmë e madhe në përmirësimin e aftësive motorike, forcës dhe koordinimit. Kjo terapi është ndihmë e madhe që fëmija të jetë në gjendje të kryejë aktivitetet e përditshme. Në rastet e anomalive të rënda fizike, të tilla si deformime të gjymtyrëve apo gjoksit, mund të kryhen edhe ndërhyrjet operative nga psikologët për të përballuar sfidat dhe për të përmirësuar vetëvlerësimin dhe aftësitë sociale të fëmijës. Fëmijët mund të kenë nevojë për një program të specializuar arsimor ose përshtatje shkollore.