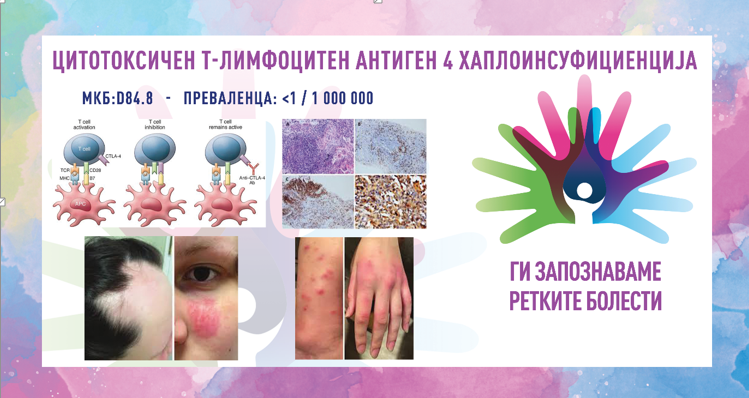
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
КАРНИЕВ КОМПЛЕКС/БЕНИГНА НЕОПЛАЗМА НА ЕНДОКРИНИТЕ ЖЛЕЗДИ / CARNEY COMPLEX (CNC)/COMPLEXUS CARNEY
MKБ-10 D44.8
ПРЕВАЛЕНЦА – Карниевиот комплекс е многу ретко заболување, па затоа податоците во поглед на преваленцата се ограничени, но се претпоставува дека се јавува 1/1 000.000 население (приближно 750 пријавени и опишани случаи во литературата).
ВОВЕД
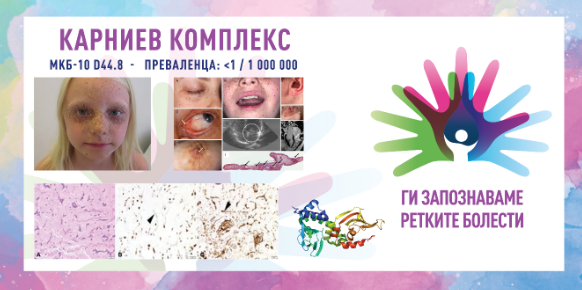
Карниевиот комплекс (Carney complex) е редок мултисистемски генетски синдром што најчесто се манифестира со појава на бенигни тумори, пигментни промени на кожата и нарушувања во функцијата на ендокрините жлезди. Синдромот е именуван по д-р Џ. А. Карни, кој прв го опишал овој комплекс во 1985 година. Поради неговите разновидни клинички манифестации, Карниевиот комплекс се смета за важен дијагностички предизвик и во овој случај се јавува потреба од мултидисциплинарен пристап во лекувањето. Во повеќето случаи, Карниевиот комплекс се наследува на автосомно доминантен начин, што значи дека е доволна само една мутирана копија на генот за да се појави синдромот, но можни се и спорадични случаи без семејна историја.
ЕТИОЛОГИЈА
Во повеќе од 70% од случаите, Карниевиот комплекс е поврзан со мутации на генот PRKAR1A кој ја кодира регулаторната подединица R1alpha на протеин киназа А. Мутацијата c.709-7del6 е главно поврзана со изолирана PPNAD, а мутацијата c.491-492del е поврзана со почести срцеви миксоми, лентигини и тумори на тироидната жлезда. Опишани се и штетни варијанти на PDE11A кај пациенти со PRKAR1A-мутации, со почести PPNAD и тумори на тестисите. Мутација на трипликација на герминативните линии што вклучува PRKACB, генот што ја кодира каталитичката подединица бета на протеинот киназа А, е опишана кај млада жена со акромегалија, лентигини и миксоми која не поседувала мутација на PRKAR1A.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Клиничката слика на Карниевиот комплекс е многу разновидна и може да се разликува меѓу пациентите, но најчестите симптоми вклучуваат:
кожни промени – хиперпигментирани дамки, како кафеави макули (кафеава боја) или сини невуси, кои најчесто се појавуваат на лицето, вратот и рацете;
миксоми – овие се бенигни тумори што можат да се појават на кожата, срцето (особено атриумите), дојките или мукозните мембрани; срцевите миксоми се доста опасни бидејќи можат да доведат до опструкција на срцевите комори, тромбоза или емболија;
ендокрини тумори и нарушувања – надбубрежни тумори што доведуваат до Кушингов синдром поради прекумерна продукција на кортизол; пациентите можат да имаат и акромегалија (прекумерен раст на коските и ткивата) и дисфункција на тироидната жлезда;
тумори на тестисите и овариумите – тие можат да се појават како пигментни нодули и да доведат до нарушување на репродуктивните функции;
неоплазии на централниот нервен систем – иако ретко, можат да се појават тумори на хипофизата или на други делови од мозокот.
Дијагностицирањето се прави врз основа на присуството на двете главни карактеристики (периорфикални летигини, миксоми или PPNAD) или една главна карактеристика со дополнителен критериум, како што е мутација на PRKAR1A или засегнат роднина од прв степен. Пациентите со Карниев комплекс, или со генетска предиспозиција за Карниевиот комплекс треба да имаат редовен скрининг за манифестации на болеста, најмалку еднаш годишно кај сите пациенти. Сепак, при дијагностицирањето треба да се внимава и на диференцијалните дијагнози, особено во случаите кога PPNAD може да се изолира без други манифестации на Карниевиот комплекс и без генетска варијанта на PRKAR1A, или да биде дел од класичен Карниев комплекс поврзан со друга главна карактеристика, како лентигиноза и/или миксом.
ПОСЛЕДИЦИ
Последиците од Карниевиот комплекс се различни и зависат од тоа кои органи се погодени. Срцевите миксоми можат да предизвикаат сериозни компликации, како што се срцеви блокади, аритмии и срцева слабост, кои можат да бидат опасни по живот. Надбубрежните тумори и хормоналните нарушувања можат да доведат до метаболитички нарушувања и зголемен ризик од дебелина, дијабетес и хипертензија. Долгорочната прогноза зависи од раното откривање и успешноста на третманот на туморите и хормоналните нарушувања.
ТРЕТМАН
При лекувањето на Карниевиот комплекс се јавува потреба од индивидуален пристап, кој вклучува:
хируршка интервенција – отстранување на миксомите и другите тумори; за срцевите миксоми, операцијата е приоритетна во поглед на спречување на срцевите компликации;
мониторинг и дијагностички тестови – редовни ултразвучни прегледи, ехокардиографии, МРИ и компјутерски томографии за следење на растот и развојот на туморите и другите абнормалности;
ендокринолошка терапија – за пациентите со ендокрини нарушувања, третманот може да вклучува лекови за регулирање на хормоните и намалување на симптомите, како и супресивна терапија за некои ендокрини тумори;
билатералната адреналектомија – најчестиот третман поради PPNAD;
генетско советување – околу 70 % од случаите се фамилијарни со автосомно доминантен пренос, па затоа за пациентите и нивните семејства од голема важност се процената на ризикот од наследување и поддршката во планирањето на семејството.
Редовната медицинска грижа и мултидисциплинарниот пристап овозможуваат подобра контрола на симптомите и подобрување на квалитетот на животот на пациентите со Карниевиот комплекс.
KOMPLEKSI CARNEY/NEOPLAZMA BENIJE E GJENDRAVE ENDOKRINE / CARNEY COMPLEX (CNC)/COMPLEXUS CARNEY
ICD-10 D44.8
PREVALENCA – Kompleksi i Carney është sëmundje shumë e rrallë, kështu që të dhënat e prevalencës janë të kufizuara, por supozohet se ndodh në 1/1,000,000 popullatë (afërsisht 750 raste të raportuara dhe të përshkruara në literaturë).
HYRJE
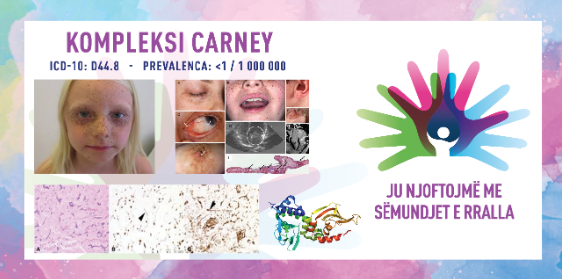
Kompleksi Carney është sindromë e rrallë gjenetike multisistemike që më së shpeshti manifestohet me shfaqjen e tumoreve beninje, ndryshime pigmentare në lëkurë dhe çrregullime në funksionin e gjëndrave endokrine. Sindroma është emëruar sipas Dr. J. A. Carney, i cili e përshkroi për herë të parë këtë kompleks në vitin 1985. Për shkak të manifestimeve të ndryshme klinike, kompleksi Carney konsiderohet një sfidë e rëndësishme diagnostike dhe në këtë rast ka nevojë për një qasje multidisiplinare në trajtim. Në shumicën e rasteve kompleksi Carney trashëgohet në mënyrë autosomale dominante, që do të thotë se mjafton vetëm një kopje e mutuar e gjenit që të shfaqet sindroma, por janë të mundshme edhe raste sporadike pa histori familjare.
ETIOLOGJIA
Në më shumë se 70% të rasteve kompleksi Carney shoqërohet me mutacione në gjenin PRKAR1A, i cili kodon nën-njësinë rregullatore R1alfa të proteinës kinazës A. Mutacioni c.709-7del6 shoqërohet kryesisht me PPNAD të izoluar, ndërsa mutacioni c.491-492del shoqërohet me miksoma kardiake më të shpeshta, lentiginat dhe tumoret e tiroidës. Variantet e dëmshme të PDE11A janë përshkruar gjithashtu te pacientët me mutacione PRKAR1A, me PPNAD më të shpeshta dhe tumore testikulare. Një mutacion i trefishimit të linjës germinale që përfshin PRKACB, gjen i cili kodon nën-njësinë beta katalitike të proteinës kinazë A, është përshkruar te një grua e re me akromegali, lentigina dhe miksoma që nuk kishte një mutacion PRKAR1A.
SIMPTOMAT DHE DIAGNOSTIKIMI
Pamja klinike e kompleksit Carney është shumë e larmishme dhe mund të ndryshojë midis pacientëve, por simptomat më të zakonshme përfshijnë:
ndryshimet e lëkurës – njolla të hiperpigmentuara, si makula me ngjyrë kafe ose nevi blu, të cilat zakonisht shfaqen në fytyrë, qafë dhe duar;
miksomat – këto janë tumore beninje që mund të shfaqen në lëkurë, në zemër (veçanërisht në atrium), në gjoks ose në mukozë; miksomat kardiake janë mjaft të rrezikshme sepse mund të çojnë në bllokim të dhomave të zemrës, trombozë ose emboli;
tumoret dhe çrregullimet endokrine – tumoret mbiveshkore që çojnë në sindromën Cushing për shkak të prodhimit të tepërt të kortizolit; pacientët mund të kenë gjithashtu akromegali (rritje e tepërt e kockave dhe indeve) dhe mosfunksionim të tiroidës;
tumoret e testikujve dhe vezoreve – ato mund të shfaqen si nyje pigmenti dhe të çojnë në ndërprerje të funksioneve riprodhuese;
neoplazitë e sistemit nervor qendror – edhe pse rrallë mund të shfaqen tumore të gjëndrës së hipofizës ose pjesëve të tjera të trurit.
Diagnoza bazohet në praninë e të dy tipareve kryesore (letigina periorfike, miksoma ose PPNAD) ose një tipar kryesor me kriter shtesë, si p.sh. një mutacion PRKAR1A ose një i afërm i prekur i shkallës së parë. Pacientët me kompleksin Carney, ose me një predispozicion gjenetik ndaj kompleksit Carney duhet të kenë kontroll të rregullt për manifestimet e sëmundjes, të paktën një herë në vit për të gjithë pacientët. Megjithatë, gjatë diagnostikimit duhet kushtuar vëmendje edhe diagnozave diferenciale, veçanërisht në rastet kur PPNAD mund të izolohet pa manifestime të tjera të kompleksit Carney dhe pa një variant gjenetik të PRKAR1A, ose të jetë pjesë e kompleksit klasik Carney të lidhur me një tjetër tipar kryesor, të tilla si lentiginoza dhe/ose miksoma.
PASOJAT
Pasojat e kompleksit Carney janë të ndryshme dhe varen nga organet që preken. Miksoma kardiake mund të shkaktojë komplikime serioze, siç janë bllokimet e zemrës, aritmi dhe dështimi i zemrës, të cilat mund të jenë kërcënuese për jetën. Tumoret e veshkave dhe çrregullimet hormonale mund të çojnë në çrregullime metabolike dhe rrezik në rritje të obezitetit, diabetit dhe hipertensionit. Prognoza afatgjatë varet nga zbulimi i hershëm dhe trajtimi i suksesshëm i tumoreve dhe çrregullimeve hormonale.
TRAJTIMI
Gjatë trajtimit të kompleksit Carney ekziston nevoja për një qasje individuale, e cila përfshin:
ndërhyrje kirurgjikale – heqje e miksomave dhe tumoreve të tjera; për miksomat kardiake, operacioni është prioritet për parandalimin e komplikimeve kardiake;
teste monitoruese dhe diagnostike – ekzaminime të rregullta me ultratinguj, ekokardiografi, MRI dhe tomografi kompjuterike për të monitoruar rritjen dhe zhvillimin e tumoreve dhe anomalitë e tjera;
terapi endokrinologjike – për pacientët me çrregullime endokrine, trajtimi mund të përfshijë barna për rregullimin e hormoneve dhe reduktimin e simptomave, si dhe terapi supresive për disa tumore endokrine;
adrenalektomi bilaterale – trajtim më i zakonshëm për PPNAD
këshillim gjenetik – rreth 70% e rasteve janë familjare me transmetim autosomal-dominues, prandaj për pacientët dhe familjet e tyre, vlerësimi i rrezikut të trashëgimisë dhe mbështetja në planifikimin familjar kanë rëndësi të madhe.
Kujdesi i rregullt mjekësor dhe qasja multidisiplinare mundësojnë kontroll më të mirë të
simptomave dhe përmirësim të cilësisë së jetës së pacientëve me kompleksin Carney.