Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
ЛИПОДИСТРОФИЈА/ LIPODYSTROPHY
MKБ-10 Е88.1
ПРЕВАЛЕНЦА
Проценката на преваленцата за дијагностицирана липодистрофија е 2,8 случаи на милион луѓе. Конгениталната липодистрофија (поради наследен генетски дефект) се проценува дека е исклучително ретка, а веројатно влијае само на еден на милион луѓе. Една подоцнежна студија објавена во изминатата година сугерирала клиничка преваленца на липодистрофија со повисока тенденција, и тоа до 1 на 20 000 индивидуи и генетска форма со тенденција до 1 на 7 000, особено кај лицата со ХИВ-инфекција.
ВОВЕД
Синдромите на липодистрофија се група генетски или стекнати нарушувања во кои телото не е во состојба да произведе и одржува здраво масно ткиво. Медицинската состојба се карактеризира со абнормални или дегенеративни состојби на масното ткиво на телото. Поконретно, липоатрофија (од грчки липо – „масти“ и дистрофија – „абнормална или дегенеративна состојба“) се користи кога се опишува губењето на маснотии од една област (обично лицето). Оваа состојба се карактеризира и со недостиг на циркулирачки лептин што може да доведе до остеосклероза. Отсуството на масно ткиво е поврзано со отпорност на инсулин, хипертриглицеридемија, безалкохолна масна болест на црниот дроб (NAFLD) и метаболички синдром.
Липодистрофијата може да се подели на:
синдроми на вродена липодистрофија;
вродена генерализирана липодистрофија (синдром Berardinelli–Seip);
семејна делумна липодистрофија;
Марфаноид-прогероид-липодистрофија;
хронична атипична неутрофилна дерматоза со липодистрофија и синдром на покачена температура;
стекнати синдроми на липодистрофија;
стекната делумна липодистрофија (Баракер–Симонсов синдром);
стекната генерализирана липодистрофија;
центрифугална абдоминална липодистрофија (Lipodystrophia centrifugalis abdominalis infantilis);
прстенеста липоатрофија (Ferreira-Marques lipoatrophia);
локализирана липодистрофија;
липодистрофија поврзана со ХИВ.
ЕТИОЛОГИЈА
Генетските форми на липодистрофија, конгенитална генерализирана липодистрофија и семејна парцијална липодистрофија, се предизвикани од одредени генетски мутации (промени). Мутациите во гените AGPAT2, BSCL2, CAV1 и CAVIN1 предизвикуваат конгенитална генерализирана липодистрофија тип 1, 2, 3, 4, соодветно. Овие гени играат важна улога во развојот и функцијата на адипоцитите, клетки што складираат маснотии во човековото масно ткиво. Мутациите на овие гени влијаат на структурата и функцијата на адипоцитите. Семејната делумна липодистрофија може да биде предизвикана од мутации на неколку гени, најчесто мутации во генот LMNA. Дете со CGL ги наследува генските мутации од неговите биолошки родители. Секој од родителите носи по една копија од мутираниот ген, но тие обично не покажуваат знаци и симптоми на состојбата. Стекнатите липодистрофии можат да бидат предизвикани од лекови, автоимуни реакции или да имаат непозната причина (идиопатска). Додека стекнатите липодистрофии немаат директна генетска основа, некои истражувачи мислат дека некои луѓе можат да имаат генетска предиспозиција за развој на одредени форми на стекната липодистрофија.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
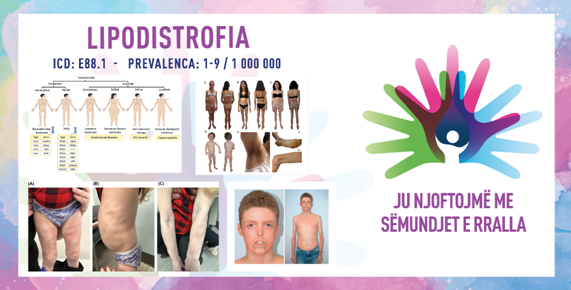
Бидејќи има многу видови на липодистрофија, нејзините симптоми варираат во голема мера, па засегнатите можат да немаат здравствени проблеми со години. Најчестиот симптом на липодистрофија е забележливо и постојано намалување на количината на маснотии во некои делови од телото или релативно вишок маснотии во други делови од телото. Луѓето со стекната делумна липодистрофија постепено губат маснотии од лицето, вратот, рацете и градите во текот на детството. Некои луѓе со APL можат да имаат вишок маснотии околу стомакот, нозете или задникот. Оние кои имаат суптилен поттип на синдромот можат да покажат необично и необјасниво високи нивоа на холестерол, триглицериди и гликоза при рутински тестови на крвта. Симптомите, исто така, се движат по сериозност, од ненаситен апетит на едниот крај, до неподнослива болка поради панкреатитис (воспаление на панкреасот) на другиот крај. Друг знак може да биде ако пациентот страда од хипергликемија (премногу шеќер во крвта) или хипертриглицеридемија (премногу триглицериди во крвта) која е отпорна на нормален третман. Дијагностицирањето вообичаено е врз основа на клиничка слика, физички преглед, семејна историја на медицински состојби и искуство на ендокринолог. Користењето дебеломер за преклопување на кожата за мерење на дебелината на наборите на кожата во различни делови од телото или скенирање на вкупниот состав на телото со помош на Двојна енергетска рендгенска апсорптиометрија, исто така, може да помогне да се идентификува поттипот. Апсорптиометријата со рендген со двојна енергија може да биде корисна со обезбедување на регионални мерења на % масти и директна визуелизација на дистрибуцијата на маснотиите со помош на „масна сенка“. Понекогаш генетската потврда е можна во зависност од поттипот со помош на Next-Generation Sequencing (NGS) Gene Panels и Whole-Exome Sequencing (WES) генетско тестирање. Сепак, кај до 40 % од пациентите со делумна липодистрофија, не е идентификуван предизвикувачкиот ген.
ПОСЛЕДИЦИ
Поради недоволниот капацитет на поткожното ткиво да складира маснотии, маснотиите се депонираат во немасното ткиво (липотоксичност), што доведува до отпорност на инсулин. Пациентите можат да покажат хипертриглицеридемија, тешка масна болест на црниот дроб, и делумно или никакво масно ткиво. Просечниот животен век на пациентот е приближно 30 години, со откажување на црниот дроб како вообичаена причина за смрт. За разлика од високите нивоа забележани кај безалкохолната масна болест на црниот дроб (NAFLD) поврзана со дебелината, кај липодистрофијата, нивоата на лептин се многу ниски.
ТРЕТМАН
Третманот на липодистрофијата зависи од тоа за кој специфичен тип се работи и дали некои други состојби се поврзани со веќе постоечката, како што се дијабетес или абнормални нивоа на холестерол.
1. модификација на животниот стил (диета и вежбање по потреба), метформин и фибрати (и/или статини);
2. метформин, тиазолидиндиони – во третман на инсулинска резистенција;
3. фибрати (и/или статини) – во третман на дислипидемија (хиперхолестеролемија);
4. козметички третман, естетка пластична хирургија;
5. баријатриска хирургија (Roux-en-Y гастричен бајпас хирургија/ RYGB);
Терапијата за замена на лептин со хуман рекомбинантен лептин метрелептин се покажала како ефикасна терапија за ублажување на метаболичките компликации поврзани со липодистрофијата и е одобрена од FDA за третман на синдроми на генерализирана липодистрофија. Во Европа врз основа на ЕМА, метрелептинот треба да се користи како додаток на диетата за лекување на липодистрофија, при што пациентите имаат губење на масното ткиво под кожата и таложење на маснотиите на други места во телото, како што се црниот дроб и мускулите. Лекот се користи кај: возрасни и деца на возраст над две години со генерализирана липодистрофија (синдром на Berardinelli–Seip и Лоренсов синдром) и кај возрасни и деца на возраст над 12 години со делумна липодистрофија (вклучувајќи и синдром на Barraquer–Simons), во случаи кога стандардните третмани се безуспешни. Volanesorsen е Apo-CIII инхибитор и е лек кој моментално се истражува како потенцијален терапевтски лек за намалување на нивото на хипертриглицериди кај пациенти со фамилијарна парцијална липодистрофија во студијата BROADEN.
LIPODISTROFIA / LIPODYSTROPHY
ICD-10 E88.1
PREVALENCA – Prevalenca e vlerësuar e lipodistrofisë së diagnostikuar është 2,8 raste për një milion banorë. Lipodistrofia kongjenitale (si pasojë e një defekti gjenetik të trashëguar) vlerësohet të jetë jashtëzakonisht e rrallë dhe me shumë gjasë prek vetëm 1 në një milion njerëz. Një studim më i fundit, i publikuar gjatë vitit të kaluar, sugjeron një prevalencë klinike më të lartë të lipodistrofisë, deri në 1 në 20 000 individë, dhe të formës gjenetike deri në 1 në 7 000, veçanërisht te personat me infeksion HIV.
HYRJE
Sindromat e lipodistrofisë përbëjnë një grup çrregullimesh gjenetike ose të fituara, në të cilat trupi nuk është në gjendje të prodhojë dhe të ruajë ind dhjamor të shëndetshëm. Kjo gjendje mjekësore karakterizohet nga ndryshime anormale ose degjenerative të indit dhjamor të trupit.
Më konkretisht, lipoatrofia (nga greqishtja lipo – “yndyrë” dhe distrofi – “gjendje anormale ose degjenerative”) përdoret për të përshkruar humbjen e yndyrës nga një zonë e trupit (zakonisht fytyra). Kjo gjendje karakterizohet gjithashtu nga mungesa e leptinës qarkulluese, e cila mund të çojë në osteosklerozë. Mungesa e indit dhjamor shoqërohet me rezistencë ndaj insulinës, hipertrigliceridemi, sëmundje të mëlçisë yndyrore jo-alkoolike (NAFLD) dhe sindrom metabolik.
Lipodistrofia mund të ndahet në:
sindroma të lipodistrofisë kongjenitale;
lipodistrofi kongjenitale e gjeneralizuar (sindroma Berardinelli–Seip);
lipodistrofi familjare e pjesshme;
lipodistrofi marfanoide–progeroide;
dermatozë kronike atipike neutrofile me lipodistrofi dhe sindromë febrile;
sindroma të fituara të lipodistrofisë;
lipodistrofi e fituar e pjesshme (sindroma Barraquer–Simons);
lipodistrofi e fituar e gjeneralizuar;
lipodistrofi abdominale centrifugale (Lipodystrophia centrifugalis abdominalis infantilis);
lipoatrofi unazore (Ferreira-Marques lipoatrophia);
lipodistrofi e lokalizuar;
lipodistrofi e lidhur me HIV.
ETIOLOGJIA
Format gjenetike të lipodistrofisë, lipodistrofia kongjenitale e gjeneralizuar dhe lipodistrofia familjare e pjesshme, shkaktohen nga mutacione (ndryshime) specifike gjenetike. Mutacionet në gjenet AGPAT2, BSCL2, CAV1 dhe CAVIN1 shkaktojnë lipodistrofi kongjenitale të gjeneralizuar të tipit 1, 2, 3 dhe 4, përkatësisht. Këto gjene luajnë një rol të rëndësishëm në zhvillimin dhe funksionin e adipociteve, qelizat që ruajnë yndyrën në indin dhjamor të njeriut. Mutacionet në këto gjene ndikojnë në strukturën dhe funksionin e adipociteve.
Lipodistrofia familjare e pjesshme mund të shkaktohet nga mutacione në disa gjene, më shpesh nga mutacione në gjenin LMNA. Një fëmijë me CGL i trashëgon mutacionet gjenetike nga prindërit biologjikë; secili prind mbart një kopje të gjenit të mutuar, por zakonisht nuk shfaq shenja apo simptoma të sëmundjes.
Lipodistrofitë e fituara mund të shkaktohen nga barna, reaksione autoimune ose mund të kenë shkak të panjohur (idiopatik). Edhe pse lipodistrofitë e fituara nuk kanë bazë gjenetike direkte, disa studiues mendojnë se disa individë mund të kenë predispozicion gjenetik për zhvillimin e formave të caktuara të lipodistrofisë së fituar.
SIMPTOMAT DHE DIAGNOSTIKIMI
Duke qenë se ekzistojnë shumë lloje të lipodistrofisë, simptomat e saj ndryshojnë ndjeshëm dhe personat e prekur mund të mos kenë probleme shëndetësore për vite me radhë. Simptoma më e shpeshtë është zvogëlimi i dukshëm dhe i vazhdueshëm i sasisë së yndyrës në disa pjesë të trupit ose, përkundrazi, tepricë relative e yndyrës në pjesë të tjera.
Personat me lipodistrofi të fituar të pjesshme gradualisht humbin yndyrën nga fytyra, qafa, krahët dhe kraharori gjatë fëmijërisë. Disa persona me APL mund të kenë tepricë yndyre rreth barkut, këmbëve ose vitheve. Ata që kanë një nën-tip më të butë të sindromës mund të shfaqin nivele të pashpjegueshme dhe anormalisht të larta të kolesterolit, triglicerideve dhe glukozës gjatë analizave rutinë të gjakut.
Ashpërsia e simptomave varion nga oreks i pangopur në njërin ekstrem, deri te dhimbje të padurueshme për shkak të pankreatitit (inflamacion i pankreasit) në tjetrin. Një shenjë tjetër mund të jetë hiperglicemia (sheqer i lartë në gjak) ose hipertrigliceridemia (trigliceride të larta në gjak) rezistente ndaj trajtimit standard.
Diagnostikimi zakonisht bazohet në pamjen klinike, ekzaminimin fizik, historinë familjare mjekësore dhe përvojën e endokrinologut. Përdorimi i kaliperit për matjen e trashësisë së palave të lëkurës në zona të ndryshme të trupit, ose skanimi i përbërjes totale trupore me absorptiometri me rreze X me energji të dyfishtë (DEXA), mund të ndihmojë në identifikimin e nën-tipit. Absorptiometri me rreze X me energji të dyfishtë ofron matje rajonale të përqindjes së yndyrës dhe vizualizim të drejtpërdrejtë të shpërndarjes së saj përmes “hijes yndyrore”.
Në disa raste, konfirmimi gjenetik është i mundur në varësi të nën-tipit, duke përdorur Next-Generation Sequencing (NGS) Gene Panels dhe Whole-Exome Sequencing (WES). Megjithatë, te deri në 40% e pacientëve me lipodistrofi të pjesshme, nuk identifikohet gjen shkaktar.
PASOJAT
Për shkak të kapacitetit të pamjaftueshëm të indit nënlëkuror për të ruajtur yndyrën, ajo depozitohet në indet jo-dhjamore (lipotoksicitet), duke çuar në rezistencë ndaj insulinës. Pacientët mund të shfaqin hipertrigliceridemi, sëmundje të rëndë të mëlçisë yndyrore dhe mungesë të pjesshme ose totale të indit dhjamor. Jetëgjatësia mesatare e pacientëve është rreth 30 vjet, me insuficiencë hepatike si shkak të shpeshtë të vdekjes. Ndryshe nga nivelet e larta të leptinës të vërejtura në NAFLD (sëmundje të mëlçisë yndyrore jo-alkoolike) të lidhur me obezitetin, në lipodistrofi nivelet e leptinës janë shumë të ulëta.
TRAJTIMI
Trajtimi i lipodistrofisë varet nga tipi specifik dhe nga prania e gjendjeve të tjera shoqëruese, si diabeti ose nivelet anormale të kolesterolit.
1. modifikimi i stilit të jetesës (dietë dhe aktivitet fizik sipas nevojës), metforminë dhe fibrate (dhe/ose statina);
2. metforminë, tiazolidinedione – për trajtimin e rezistencës ndaj insulinës;
3. fibrate (dhe/ose statina) – për trajtimin e dislipidemisë (hiperkolesterolemisë);
4. trajtim kozmetik, kirurgji plastike estetike;
5. kirurgji bariatrike (bypass gastrik Roux-en-Y / RYGB).
Terapia zëvendësuese me leptinë, me leptinë humane rekombinante metreleptinë, është treguar efektive në zbutjen e komplikimeve metabolike të lidhura me lipodistrofinë dhe është miratuar nga FDA për trajtimin e sindromave të lipodistrofisë së gjeneralizuar. Në Evropë, sipas EMA-s, metreleptina përdoret si shtesë e dietës për trajtimin e lipodistrofisë, kur pacientët kanë humbje të indit dhjamor nënlëkuror dhe depozitim të yndyrës në vende të tjera të trupit, si mëlçia dhe muskujt.
Ky ilaç përdoret te: të rriturit dhe fëmijët mbi 2 vjeç me lipodistrofi të gjeneralizuar (sindroma Berardinelli–Seip dhe sindroma Lawrence) dhe te të rriturit dhe fëmijët mbi 12 vjeç me lipodistrofi të pjesshme (përfshirë sindromën Barraquer–Simons), në rastet kur trajtimet standarde janë të pasuksesshme.
Volanesorsen është një inhibitor i Apo-CIII dhe aktualisht po studiohet si një terapi potenciale për uljen e niveleve të hipertriglicerideve te pacientët me lipodistrofi familjare të pjesshme, në studimin BROADEN.