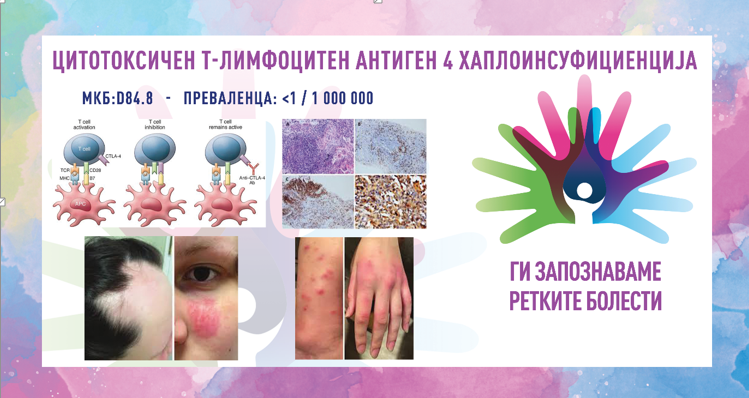
Во рамките на 8. сезона на националната кампања „Ги запознаваме ретките болести“, што ја изведува здружението „Ретко е да си редок“, секој ден од февруари ќе објавуваме по еден текст за различни ретки состојби од кои заболуваат граѓаните во Македонија.
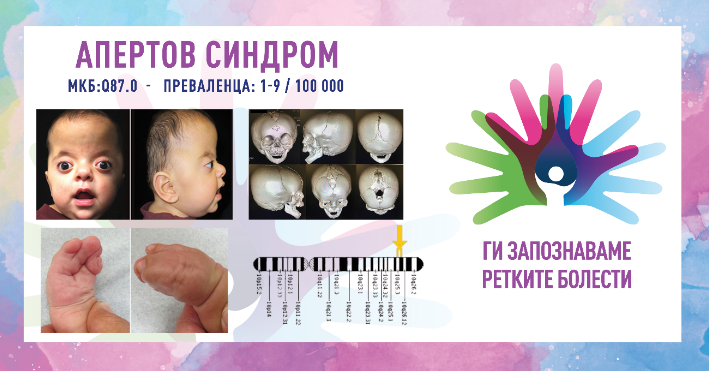
АПЕРТОВ СИНДРОМ/APERT SYNDROME
MKБ-10 Q87.0
ПРЕВАЛЕНЦА – забележана приближно кај 1/100 000 живородени деца.
ВОВЕД
Апертовиот синдром е ретко генетско нарушување кое се карактеризира со краниосиностоза (предвремена фузија на коските на черепот), абнормалности на средината на лицето и синдактилија (фузија на прстите на рацете и нозете). Состојбата е дел од групата нарушувања познати како синдроми на краниосиностоза. Апертовиот синдром обично се дијагностицира при раѓање поради неговите различни физички карактеристики и притоа се јавува потреба од мултидисциплинарна нега. Синдромот на Аперт првпат го опишал францускиот лекар Ежен Аперт, во 1906 година кога забележал уникатна комбинација на абнормалности на кранијалните нерви, лицето и рацете кај девет пациенти.
ЕТИОЛОГИЈА
Апертовиот синдром е предизвикан од мутација на генот FGFR2. Овој ген игра клучна улога во развојот и одржувањето на коските и ткивата, особено во регулирањето на растот и созревањето на клетките. Мутацијата води до абнормална сигнализација во формирањето на коските, особено во черепот и екстремитетите.
1. генетска мутација – синдромот е предизвикан од специфични мутации во генот FGFR2, кој се наоѓа на хромозомот 10; мутацијата води до предвремено спојување на кранијалните конци (краниосиностоза) и абнормален развој на коските во рацете и стапалата.
2. наследување – состојбата е обично спорадична (се јавува кај луѓе без семејна историја на нарушување), но може да се наследи и на автосомно-доминантен начин, што значи дека една копија од мутираниот ген е доволна за да ја предизвика состојбата.
3. напредна таткова возраст – постои зголемена инциденца на нови мутации кај децата родени од постари татковци, иако тоа не е единствената причина.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Симптомите на Апертовиот синдром се разновидни и влијаат на повеќе делови од телото, првенствено черепот, лицето, рацете, стапалата, а понекогаш и на други органи. Тежината на симптомите може да варира помеѓу поединци.
1. Краниосиностоза (абнормалности на черепот)
предвремена фузија на коските на черепот – раното спојување на кранијалните конци резултира со абнормална форма на черепот (акроцефалија или череп во облик на кула) и зголемен интракранијален притисок;
карактеристики на лицето – поради абнормалниот развој на черепот, поединците често имаат високо чело, рамна средина и истакнати очи (проптоза) кои можат да изгледаат широко распоредени;
хипоплазија на средното лице – неразвиеноста на средното лице може да предизвика тешкотии со дишењето и хранењето и може да доведе до опструктивна ноќна апнеа.
2. Абнормалности на рацете и стапалата
синдактилија (спој на прсти и прсти) – една од карактеристиките на Апертовиот синдром е спојувањето на коските во прстите на рацете и нозете; вообичаено, вториот, третиот и четвртиот прст на рацете и нозете се споени;
раце со мрежа или белезници – рацете можат да имаат мрежест изглед, а подвижноста на рацете и стапалата може да биде значително нарушена.
3. Невролошки симптоми
задоцнување во развојот – некои лица со Апертов синдром можат да доживеат доцнење во развојните пресвртници, како што се одење и зборување;
когнитивна функција – додека многу поединци имаат нормална интелигенција, некои можат да имаат интелектуални пречки или потешкотии во учењето, во зависност од тежината на краниосиностозата и нејзиното влијание врз развојот на мозокот.
4. Други симптоми
проблеми со забите – вообичаени се проблеми со забите, вклучувајќи гужва, малоклузија (неусогласеност на забите) и одложено никнување на забите;
губење на слухот – некои поединци можат да имаат спроводливо губење на слухот поради структурни абнормалности во ушите;
абнормалности на кожата – во некои случаи, поединците можат да имаат кожни заболувања, вклучувајќи тешки акни или хиперхидроза (прекумерно потење);
респираторни проблеми – поради структурните абнормалности на средината на лицето, може да има опструкција на дишните патишта и тешкотии со дишењето, особено за време на спиењето (апнеа при спиење).
Дијагнозата на Апертовиот синдром се заснова на комбинација на физички преглед, студии за слики и генетско тестирање.
1. Физички преглед
краниофацијални карактеристики – различниот изглед на черепот, лицето и рацете обично им овозможува на лекарите да се сомневаат во синдромот Аперт кратко време по раѓањето;
абнормалности на рацете и стапалата – синдактилија на прстите на рацете и нозете е клучна дијагностичка карактеристика што помага да се разликува Апертовиот синдром од другите синдроми на краниосиностоза.
2. Студии за слики
Х-зраци – Х-зраците на черепот, рацете и стапалата можат да помогнат да се потврди присуството на споени кранијални конци и абнормален развој на коските;
КТ или МРИ-скенови – овие модалитети на слика се користат за да се процени степенот на краниосиностоза, да се проценат какви било абнормалности на мозокот и да се планира хируршка корекција; снимањето се користи и за следење на интракранијалниот притисок и развојот на мозокот.
3. Генетско тестирање
тестирање на генот FGFR2 – молекуларното генетско тестирање може да ја потврди дијагнозата на Апертов синдром со идентификување на мутации во генот FGFR2; ова може да помогне да се разликува Апертовиот синдром од другите слични синдроми на краниосиностоза;
пренатална дијагноза – во некои случаи, пренаталното генетско тестирање и снимањето со ултразвук можат да го дијагностицираат Апертовиот синдром пред раѓањето, ако постои семејна историја или ако се откријат карактеристични абнормалности при ултразвук.
ПОСЛЕДИЦИ
Со соодветна медицинска нега и хируршка интервенција, многу лица со Апертов синдром можат да водат релативно нормален живот. Очекуваниот животен век е типично нормален, иако може да има зголемен ризик од одредени компликации. Тежината на краниосиностозата, абнормалностите на лицето и синдактилијата ќе влијаат на потребата од постојана медицинска нега и операција во текот на детството и во зрелоста. Раната хируршка корекција на кранијалните и фацијалните деформитети значително ги подобрува резултатите.
ТРЕТМАН
При Апертовиот синдром, потребен е мултидисциплинарен пристап кој вклучува краниофацијални хирурзи, неврохирурзи, педијатри, специјалисти за ортопеди и други даватели на здравствени услуги. Третманот, првенствено, е фокусиран на управување со физичките абнормалности и спречување компликации.
1. Кранијална хирургија
ремоделирање на кранијален свод – операцијата за корекција на предвременото спојување на коските на черепот обично се изведува во детството за да се олесни зголемениот интракранијален притисок, да се подобри растот на мозокот и да се нормализира обликот на черепот;
унапредување на фронто-орбиталот – оваа операција го поместува челото и горните орбити на очите кон напред, подобрувајќи ја симетријата на лицето и спречувајќи проблеми со видот;
напредување на средината на лицето – подоцна во детството, може да се изврши операција за да се доведе средното лице напред, да се подобри дишењето, јадењето и функцијата на говорот, како и целокупниот изглед на лицето.
2. Хирургија на раце и стапала
синдактилско ослободување – операцијата често се изведува за да се одвојат споените прсти и прсти; ова ја подобрува функцијата на рацете, овозможувајќи подобро разбирање и фина контрола на моторот; при развојот на детето можат да бидат потребни повеќе операции;
ортопедски менаџмент – потребна е постојана ортопедска нега за да се решат сите функционални оштетувања на рацете и нозете и да се обезбеди правилен развој на екстремитетите.
3. Ортодонтска и стоматолошка нега
ортодонтски третман – кај поединците со Апертов синдром често има потреба од ортодонтска интервенција за да се решат стоматолошки натрупани проблеми, малоклузија и други проблеми поврзани со вилицата;
редовна грижа за забите – раните и чести стоматолошки проценки се важни за следење на развојот на забите и решавање на какви било проблеми со забите.
4. Говорна и јазична терапија
говорна терапија – поради хипоплазија на средното лице и можна опструкција на дишните патишта, некои лица со Апертов синдром можат да имаат потешкотии со говорот; раната интервенција со говорна и јазична терапија може да помогне во подобрување на комуникациските вештини.
5. Управување со слухот и дишењето
слушни помагала – ако има губење на слухот, можат да бидат потребни слушни помагала за да се обезбеди правилен аудитивен развој;
управување со апнеа при спиење – ако се дијагностицира опструктивна ноќна апнеа, потребни се третмани, како континуиран позитивен притисок на дишните патишта (CPAP) или хируршка интервенција.
6. Психолошка поддршка и дефектологија
развојна поддршка – децата со доцнење во развојот можат да имаат корист од програмите за рана интервенција, од специјализираните образовни услуги и индивидуализирана поддршка за максимизирање на нивниот когнитивен и социјален развој;
семејно советување – генетското советување и психолошката поддршка за семејството се важни компоненти на сеопфатната грижа, помагајќи им на семејствата да ја разберат состојбата и да се справат со емоционалните и социјалните предизвици.
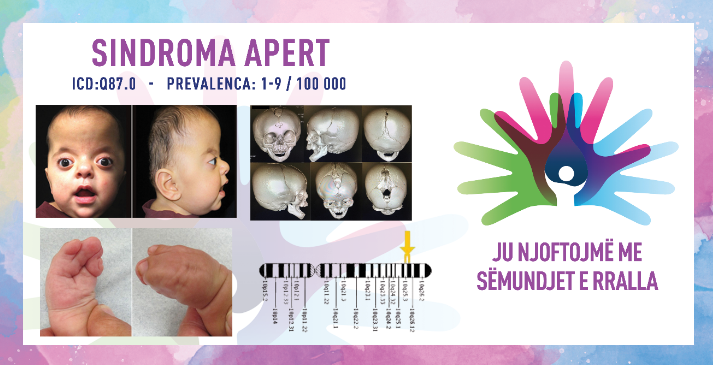
SINDROMA APERT/APERT SYNDROME
ICD-10 Q87.0
PREVALENCA – e vërejtur në afërsisht 1/100,000 lindje të gjalla.
HYRJE
Sindroma Apert është çrregullim i rrallë gjenetik që karakterizohet nga kraniosinostoza (bashkimi i parakohshëm i kockave të kafkës), abnormalitetet e mesit të fytyrës dhe sindaktilia (bashkimi i gishta të duarve dhe këmbëve). Kjo gjendje është pjesë e grupit të çrregullimeve të njohura si sindromat e kraniostenozës. Sindroma Apert zakonisht diagnostikohet menjëherë pas lindjes për shkak të karakteristikave të tij të ndryshme fizike, e për këtë arsye kërkon kujdes shumëdisiplinor. Sindromën Apert e ka përshkruar për herë të parë mjeku francez Eugèn Apert në vitin 1906, kur vërejti një kombinim unik të abnormaliteteve të nervave kraniale, fytyrës dhe duarve te nëntë pacientë.
ETIOLOGJIA
Sindroma Apert shkaktohet nga një mutacion në gjenin FGFR2. Ky gjen ka një rol kyç në zhvillimin dhe mbajtjen e kockave dhe indit, veçanërisht në rregullimin e rritjes dhe zhvillimit të qelizave. Mutacioni shkakton sinjalizim abnormal në formimin e kockave, sidomos në kafkën dhe ekstremitetet.
1. mutacioni gjenetik – sindroma shkaktohet nga mutacione të caktuara në gjenin FGFR2, i cili ndodhet në kromozomin 10; mutacioni çon në bashkimin e parakohshëm të fundeve të kafkës (kraniosinostozë) dhe zhvillim abnormal të kockave në duar dhe gishta.
2. trashëgimia – kjo gjendje është zakonisht sporadike (ndodh te individë pa histori familjare të çrregullimit), por mund të trashëgohet edhe në mënyrë autosomale dominante, që do të thotë se një kopje e gjeni të mutuar është e mjaftueshme për të shkaktuar gjendjen.
3. mosha e avancuar e babait – ka një incidencë të rritur të mutacioneve të reja te fëmijët e lindur nga baballarë më të vjetër, megjithëse kjo nuk është arsyeja e vetme.
SIMPTOMAT DHE DIAGNOSTIKIMI
Simptomat e sindromës së Apert janë të ndryshme dhe prekin pjesë të ndryshme të trupit, kryesisht kockat e kafkës, fytyrën, duart, shputat, dhe nganjëherë edhe organe të tjera. Forma e simptomave mund të ndryshojë midis individëve.
1. kraniosinostoza (anomalitë e kafkës)
përmbyllja e parakohshme e kockave të kafkës – mbyllja e parakohshme e qepallave të kafkës çon në një formë anormale të kafkës (akrocefalia ose kafkë në formë kulle) dhe rritje të presionit të intrakranial.
karakteristikat e fytyrës – për shkak të zhvillimit të parregullt të kafkës, individët shpesh kanë ballë të lartë, mes të sheshtë dhe sy të theksuar (proptoza), të cilët mund të duken si të shpërndarë shumë larg nga njëri-tjetri.
hipoplazia e fytyrës së mesme – moszhvillimi i pjesës së mesme të fytyrës mund të shkaktojë vështirësi në frymëmarrje dhe ushqim dhe mund të çojë në apne obstruktive të gjumit.
2. anomali të duarve dhe gishta
sindaktilia (bashkimi i gishta) – një nga karakteristikat e sindromës Apert është bashkimi i gishtave të duarve dhe të këmbëve; zakonisht gishti i dytë, i tretë dhe i katërt në duar dhe këmbë janë të bashkuar.
duar me rrjet ose shufra – duartë mund të kenë një pamje rrjetore, dhe lëvizshmëria e duarve dhe gishta mund të jetë e kufizuar ndjeshëm.
3. simptoma neurologjike
vonesa në zhvillim – disa individë me sindromën Apert mund të përjetojnë vonesa në arritjen e pikave të zhvillimit, siç janë ecja dhe të folurit.
funksioni kognitiv – derisa shumë individë kanë inteligjencë normale, disa mund të kenë vështirësi intelektuale ose probleme në mësim, varësisht nga shkalla e kraniosinostozës dhe ndikimi i saj në zhvillimin e trurit.
4. simptoma të tjera
probleme me dhëmbët – probleme të zakonshme me dhëmbët, përfshirë ngjeshje, malokluzi (mospërputhje e dhëmbëve) dhe vonesë në shfaqjen e dhëmbëve.
humbja e dëgjimit – disa individë mund të kenë humbje të dëgjimit për shkak të abnormaliteteve strukturore në veshë.
anomali të lëkurës – në disa raste individët mund të kenë çrregullime të lëkurës, përfshirë akne të rënda ose hiperhidrozë (djersitje e tepërt).
probleme respiratore – për shkak të abnormaliteteve strukturore të mesit të fytyrës, mund të ketë pengesa në rrugët e frymëmarrjes dhe vështirësi në frymëmarrje, veçanërisht gjatë gjumit (apnea gjatë gjumit).
Diagnoza e sindromës Apert bazohet në një kombinim të ekzaminimit fizik, studimeve të imazhit dhe testimit gjenetik.
1. Ekzaminimi fizik
karakteristikat kranio-faciale – dallimet në pamjen e kockës së kokës, fytyrës dhe duarve zakonisht lejojnë mjekët të dyshojnë për sindromën Apert herët pas lindjes.
abnormalitetet e duarëve dhe këmbëve – sindaktilia e gishtave e duarve dhe këmbëve është karakteristikë kyçe diagnostikuese që ndihmon në dallimin e sindromës Apert nga sindromat e tjera të kranio-sinostozës.
2. Studimet e imazhit
rrezet X – rrezet X të kokës, duarve dhe këmbëve mund të ndihmojnë në konfirmimin e pranisë së nyjave të mbyllura të kockave të kokës dhe zhvillimit të pazakontë të kockave.
CT ose MRI skanime – këto metoda imazherie përdoren për të vlerësuar shkallën e kranio-sinostozës, për të analizuar ndonjë abnormalitet në tru dhe për të planifikuar korrigjimin kirurgjikal; ato përdoren gjithashtu për të monitoruar presionin intracranial dhe zhvillimin e trurit.
3. Testimi gjenetik
testimi i gjeneve FGFR2 – testimi gjenetik molekular mund të konfirmojë diagnozën e sindromës Apert duke identifikuar mutacione në gjenin FGFR2; kjo mund të ndihmojë në dallimin e sindromës Apert nga sindromat e tjera të ngjashme të kranio-sinostozës.
diagnostifikimi prenatal – në disa raste testimi gjenetik prenatal dhe skanimi me ultratinguj mund ta diagnostikojnë sindromën Apert para lindjes, nëse ekziston një histori familjare ose nëse janë zbuluar abnormalitete karakteristike gjatë metodës me ultratinguj.
PASOJAT
Me kujdesin e duhur mjekësor dhe ndërhyrjet kirurgjikale shumë individë me sindromën Apert mund të jetojnë një jetë relativisht normale. Shkalla e parashikuar e jetëgjatësisë është zakonisht normale, megjithatë mund të ketë rrezik të shtuar për disa komplikime. Pesha e kraniosinostozës, abnormaliteteve të fytyrës dhe sindaktilisë do të ndikojnë në nevojën për kujdes mjekësor të vazhdueshëm dhe operacione gjatë fëmijërisë dhe në moshë të rritur. Korrektimi kirurgjik i hershëm i deformimeve kraniale dhe faciale përmirëson ndjeshëm rezultatet.
TRAJTIMI
Në sindromën Apert është e nevojshme një qasje multidisiplinare që përfshin kirurgë kranio-facialë, neurokirurgë, pediatër, specialistë ortopedikë dhe ofrues të tjerë të shërbimeve shëndetësore. Trajtimi është kryesisht i fokusuar në menaxhimin e abnormaliteteve fizike dhe parandalimin e komplikimeve.
1. Kirurgjia kraniale
rimodelimi i kupolës kraniale – operacioni për korrigjimin e bashkimit të parakohshëm të kockave të kafkës zakonisht kryhet në fëmijëri për të lehtësuar rritjen e presionit intrakranial, për të përmirësuar rritjen e trurit dhe për të normalizuar formën e kafkës.
përparimi i fronto-orbitalit – ky operacion lëviz ballin dhe orbita të sipërme të syve përpara, duke përmirësuar simetrinë e fytyrës dhe duke parandaluar problemet me shikimin.
përparimi i pjesës së mesme të fytyrës – më vonë gjatë fëmijërisë mund të kryhet operacion për të lëvizur pjesën e mesme të fytyrës përpara, për të përmirësuar frymëmarrjen, të ushqyerit dhe funksionin e të folurit, si dhe pamjen e përgjithshme të fytyrës.
2. Kirurgjia e duarve dhe këmbëve
lirimi i sindaktilisë – operacioni shpesh kryhet për të ndarë gishtat e bashkuar; kjo përmirëson funksionin e duarve, duke mundësuar një kuptim më të mirë dhe kontrollet fine të lëvizjeve; gjatë zhvillimit të fëmijës mund të kërkohen operacione të tjera.
menaxhimi ortopedik – nevojitet kujdes i vazhdueshëm ortopedik për të trajtuar çdo dëmtim funksional të duarve dhe këmbëve dhe për të siguruar zhvillimin e duhur të ekstremiteteve.
3. Kujdes ortodontik dhe stomatologjik
trajtimi ortodontik – individët me sindromën Apert shpesh kanë nevojë për ndërhyrje ortodontike për të trajtuar probleme të mbledhura stomatologjike, malokluzi dhe probleme të tjera të lidhura me nofullën.
kujdesi i rregullt i dhëmbëve – vlerësimet e hershme dhe të shpeshta stomatologjike janë të rëndësishme për të monitoruar zhvillimin e dhëmbëve dhe për të trajtuar çdo problem që mund të paraqitet me dhëmbët.
4. Terapia e të folurit dhe gjuhës
terapia e të folurit – për shkak të hipoplazisë së pjesës së mesme të fytyrës dhe mundësisë së bllokimit të rrugëve të frymëmarrjes, disa individë me sindromën Apert mund të kenë vështirësi me të folurit; ndërhyrja e hershme me terapi të të folurit dhe gjuhës mund të ndihmojë në përmirësimin e aftësive të komunikimit.
5. Menaxhimi i dëgjimit dhe frymëmarrjes
pajisje dëgjimi – nëse ka humbje të dëgjimit, mund të nevojiten pajisje dëgjimi për të siguruar zhvillimin e duhur auditiv.
menaxhimi i apnesë në gjumë – nëse diagnostikohet apne obstruktive në gjumë, janë të nevojshme trajtime, si presion i vazhdueshëm pozitiv i rrugëve të frymëmarrjes (CPAP) ose ndërhyrje kirurgjikale.
6. Mbështetje psikologjike dhe edukimi special
mbështetje zhvillimore – fëmijët me vonesa në zhvillim mund të përfitojnë nga programet e ndërhyrjes së hershme, shërbimet e edukimit të specializuar dhe mbështetje individuale për të maksimizuar zhvillimin e tyre kognitiv dhe social.
këshillimi familjar – këshillimi gjenetik dhe mbështetja psikologjike për familjen janë komponentë të rëndësishëm të kujdesit gjithëpërfshirës, duke i ndihmuar familjet të kuptojnë gjendjen dhe të përballen me sfidat emocionale dhe sociale.